生物分子结构预测领域长期面临一个两难局面:一方面,生成大量候选构象是提高预测准确性的常用策略;另一方面,无目标的大规模采样效率低下、计算成本高昂,尤其在复杂多聚体装配体中,生成的结构往往集中在局部区域,冗余多而多样性有限。研究团队提出的 HelixFold-S1 正是为解决这一困境而来——它是一种面向生物分子复合物结构预测的引导式规划方法,能够有策略地定位构象空间中信息密度最高的区域,从而生成更准确、更有价值的构象。
具体而言,对于每个复合物,模型首先预测链间接触概率,这相当于为构象空间绘制了一张粗略蓝图。有了这张图,计算资源可以集中投向高概率、低冗余的接触区域——这些接触随后作为结构生成的约束条件。跨多类生物分子复合物的基准测试表明,HelixFold-S1 相比传统无引导采样方法,显著提升了结构准确性,同时将采样需求降低了约一个数量级。更有意思的是,预测出的接触概率还能粗略指示预测难度和采样收益,相当于顺便告诉研究者“这个靶标值不值得花大力气采样”。

生物分子结构预测一直是计算生物学的核心命题,对药物发现、蛋白质工程和分子相互作用研究都至关重要。近年来,AlphaFold、RoseTTAFold 等深度学习模型确实把结构预测推到了新高度,但复杂生物分子复合物的准确预测仍然是个硬骨头。为了啃下这块骨头,研究人员尝试了各种办法:去噪多序列比对信息、整合实验约束、改进模型架构以优化相互作用界面……但在不同生物分子系统中稳定获得高精度预测,始终是悬而未决的挑战。
既然提升单次预测准确性这么难,生成大量候选构象再从中筛选高质量结果,就成了一个简单而有效的增强策略。AlphaFold 系列早就通过模型集成来提高精度,AlphaFold3 也证实大量采样可以改善蛋白质—抗体界面建模。近期的研究常常生成成百上千个候选结构来扩大构象多样性。但问题在于,这种大规模采样计算成本太高,而且当前多数采样方式缺乏明确方向,基本属于在构象空间里盲目探索。结果往往是生成的构象集中在局部区域,冗余很多,真正有用的信息增量却很有限。结构预测需要一种规划驱动的策略——先识别最可能有价值的构象区域,再把计算资源集中投进去。
HelixFold-S1 正是在这个思路下诞生的。它用引导式采样替代了传统的随机采样,第一步是预测链间残基接触概率,把它当成构象空间的粗粒度蓝图;然后选择高概率且低冗余的链间接触作为空间约束,引导结构生成。最终生成的构象根据模型置信度排序,挑出最优结构作为输出。这个策略不仅提升了准确性,还把采样成本降了下来。
方法
HelixFold-S1 基于 HelixFold3 架构搭建,加入了两个以接触为核心的新模块:接触预测模块和接触条件模块。推理过程分两个阶段。第一阶段,模型根据 Pairformer 产生的成对表示,预测链间接触概率矩阵,描述不同分子链之间哪些 token 更可能发生空间接触。第二阶段,模型按照预测概率选择接触,把这些接触作为约束输入接触条件模块,引导结构生成。
值得关注的是冗余接触剪枝策略——当某个接触约束生成了一个构象后,已经在该构象中被满足的接触会从后续候选中移除。所有生成结构再根据置信度评分排序,最高评分结构作为最终预测结果。训练时,模型在多任务框架下交替优化接触预测和接触条件结构生成,让它既能学习链间相互作用模式,也能在推理时利用这些接触约束生成更准确的复合物结构。
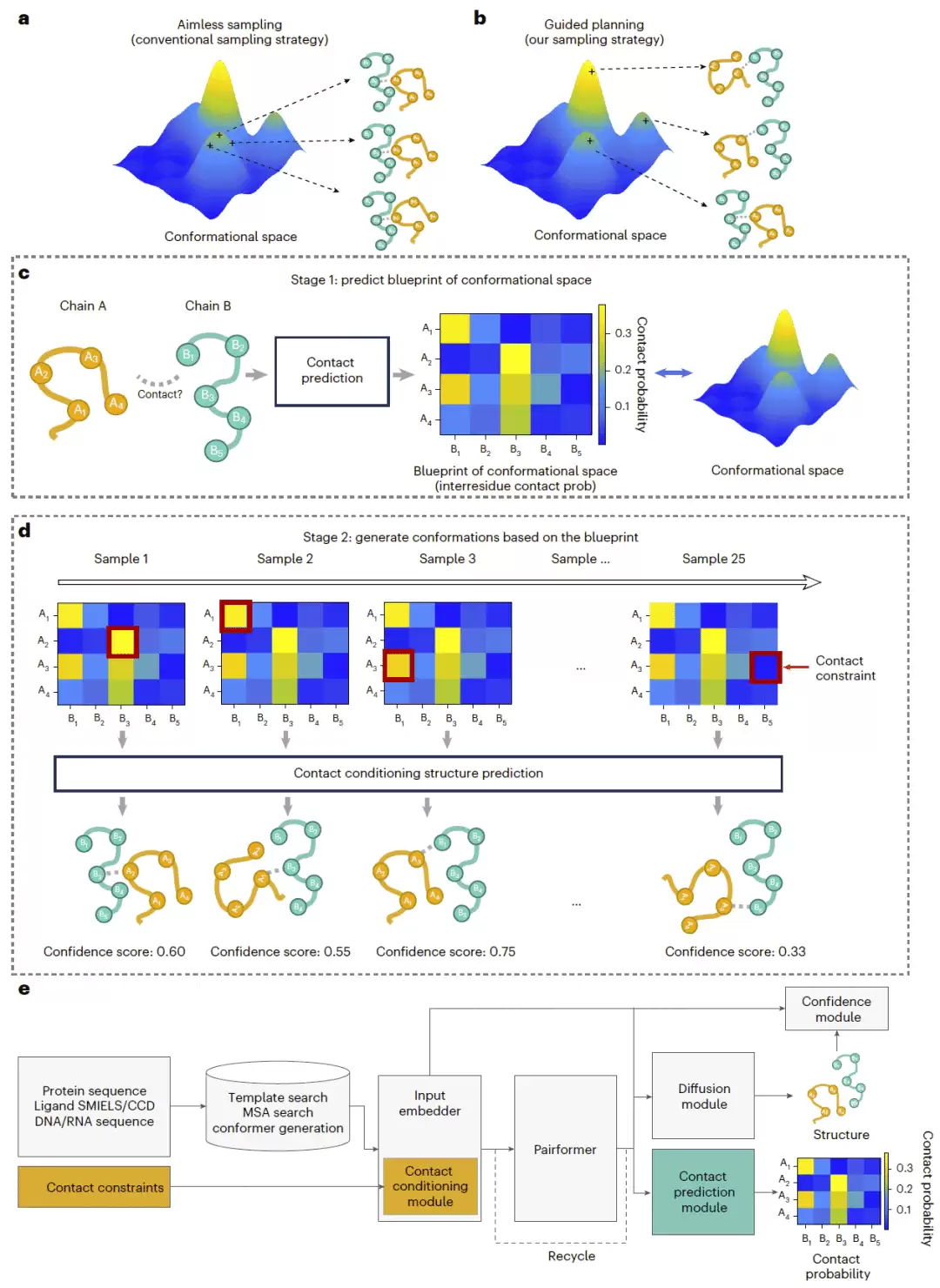
图1|HF-S1 总体框架。
结果
HF-S1 的引导式采样策略和架构
HF-S1 的核心思想说起来并不复杂:把复合物结构预测转化为“先规划、再生成”的过程。传统采样方法在构象空间里无目标搜索,很容易反复探索同一个区域;HF-S1 则先预测链间接触概率图,把它当作构象空间蓝图,识别最可能形成关键相互作用的区域。然后,模型依次选择高概率接触作为结构生成约束,优先探索那些更有可能产出正确界面的构象。接触预测模块输出链间接触概率矩阵,接触条件模块把选定的接触约束融合进结构预测过程。这样的设计让模型在更少采样次数下,生成更高质量、更多样的候选构象。
不同复合物类型中的结构准确性提升
研究团队构建了一个多类型复合物测试集,涵盖蛋白质—抗体、蛋白质—蛋白质、蛋白质—配体、蛋白质—RNA 和蛋白质—DNA 复合物,并与 HF3、HF3 结合 AFSample、Protenix、Chai-1 等方法做了对比。在每个靶标生成 25 个预测结构并按置信度排序的设置下,HF-S1 在所有复合物类型中都表现出更高的 top-5 精度。
提升最猛的是蛋白质—抗体复合物,相比 HF3 提高了超过 55%,相比 HF3 结合 AFSample 也提高了超过 33%。原因可能在于抗原结合位点的结构多样性很高,传统采样很难捕获接近天然状态的界面构象,而 HF-S1 的接触引导策略实实在在地提高了生成正确界面接触的概率。蛋白质—蛋白质、蛋白质—RNA 和蛋白质—DNA 复合物也都表现出了稳定提升。蛋白质—配体复合物的提升相对较小,可能是配体结合位点本身约束较强,预测难度相对较低。
研究团队还把 HF-S1 的接触引导策略应用到了 Protenix 和 Chai-1 上,发现不同折叠模型在加入 S1 接触约束后都能获得性能提升——这说明该策略具有一定跨模型泛化能力。在蛋白质—抗体复合物的大规模采样实验中,HF-S1 仅用 10 次采样就达到了 HF3 结合 AFSample 需要 1,000 次采样才能达到的精度水平,计算成本约为后者的 1%。在经典抗体、纳米抗体和 scFv 等不同抗体类型中,HF-S1 也都显示出了更高的 top-1 精度。
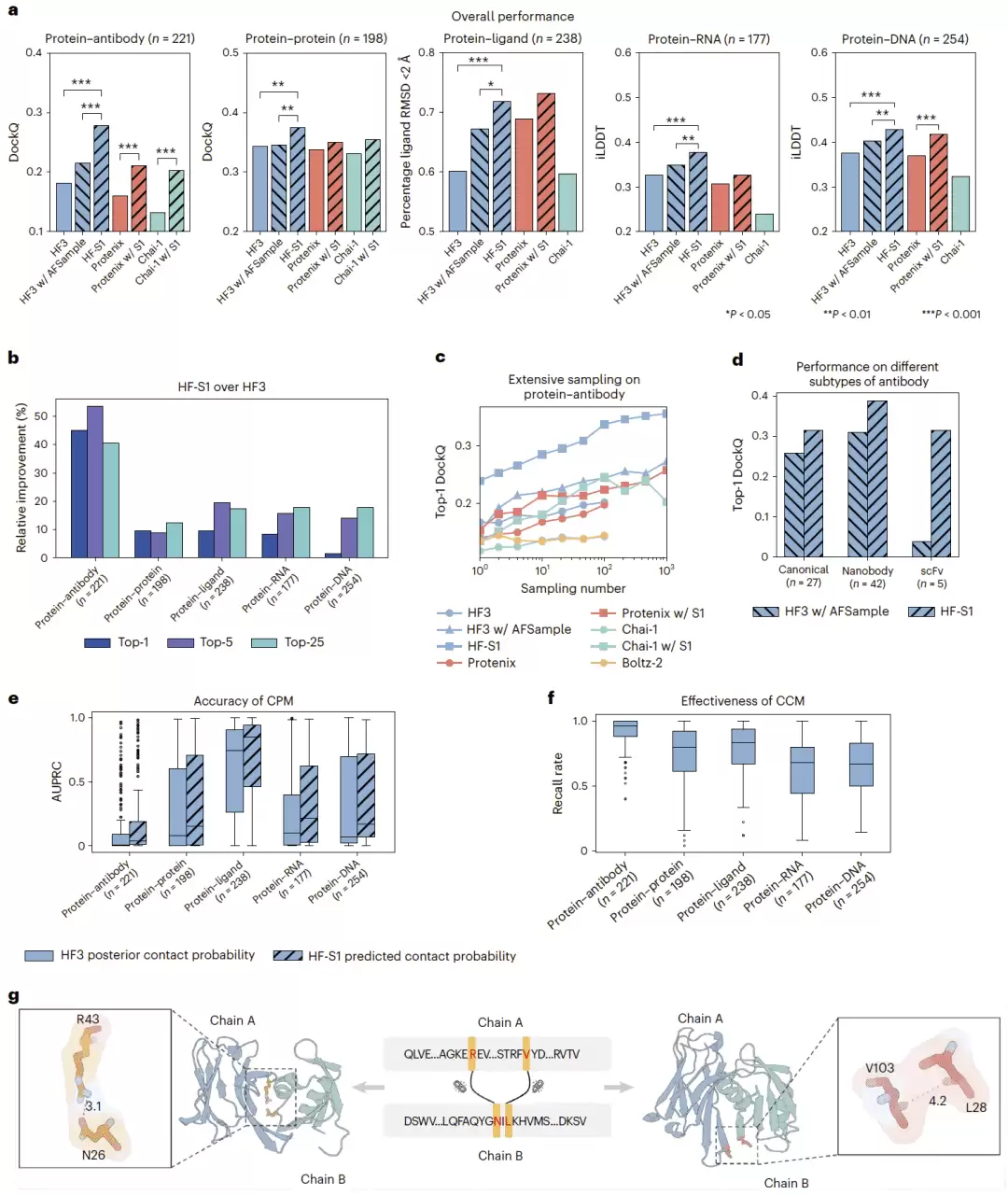
图2|HF-S1 在多类复合物中的结构预测性能和模块评估。
接触概率可指示预测难度和采样收益
HF-S1 预测的接触概率矩阵不光用于引导采样,它本身还能反映目标结构预测的内在难度。研究团队把每个靶标接触概率矩阵中的最大值定义为靶标级接触概率,然后分析它与 top-5 预测精度的关系。结果很清晰:接触概率低的靶标通常更难预测,预测精度也差;接触概率高的靶标更容易获得高精度结果。不同复合物类型中也呈现类似趋势——蛋白质—蛋白质复合物通常有比较高的接触概率,蛋白质—抗体复合物则更多集中在较低概率范围,说明它的结构不确定性更高;蛋白质—配体复合物接触概率普遍较高,说明结合位点相对容易定位。
研究团队进一步分析了接触概率与多次采样收益之间的关系。中等接触概率的靶标从多次采样中获益最大,因为它们已经有部分有用的接触信号,模型可以通过采样进一步探索和优化结构。高接触概率靶标通常一次采样就能得到不错的结构,额外采样的提升有限。低接触概率靶标虽然也能从采样中获益,但由于接触图整体信号较弱,可能需要更多采样才能找到准确构象。与 HF3 相比,HF-S1 在低接触概率靶标上的提升最大,说明引导式探索尤其适合困难目标。
此外,模型预测的残基—残基接触概率还反映了基本的理化相互作用偏好。疏水残基对如亮氨酸—亮氨酸、蛋氨酸—蛋氨酸更容易获得较高的接触概率,可能对应紧密堆积和能量有利的界面环境;而一些极性或带电残基对则倾向于较低接触概率,可能与它们的溶剂暴露或上下文依赖性相互作用有关。
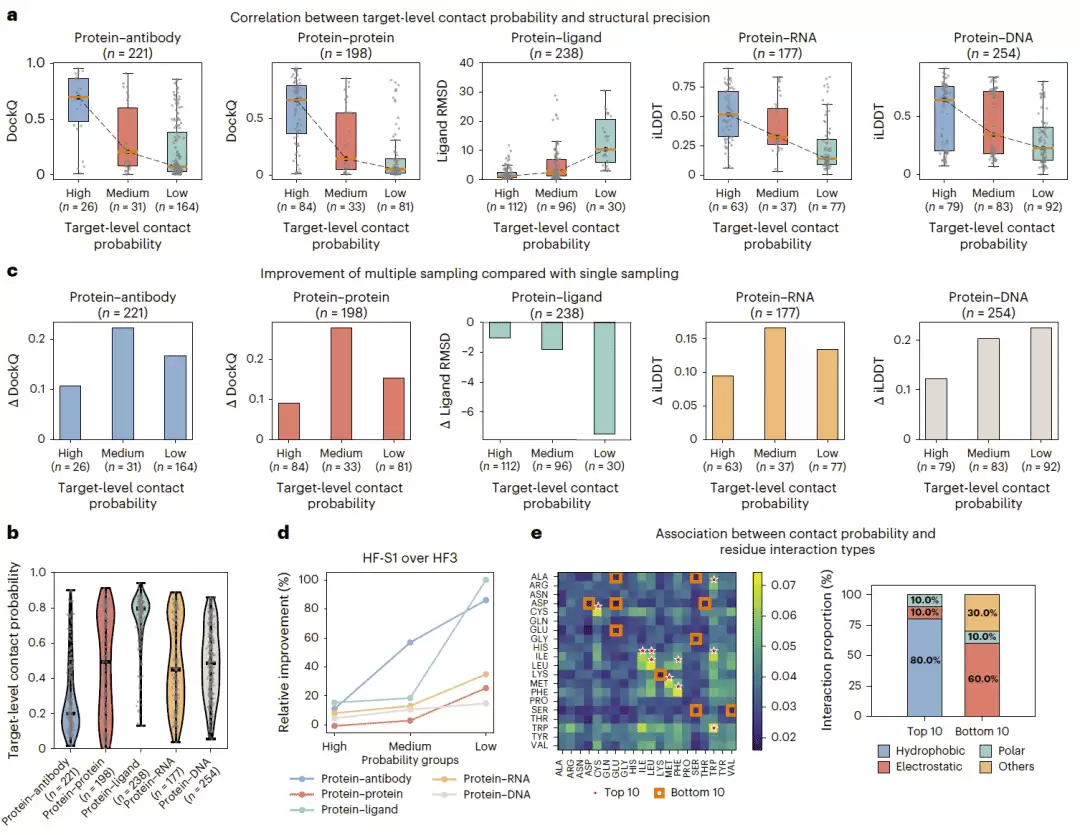
图3|接触概率作为预测难度和采样收益指标。
引导式采样改善构象空间探索
为了验证 HF-S1 是否真正改善了构象空间探索,研究团队把它和无引导采样的 HF3 做了比较,使用每个靶标采样结构精度分布的标准差作为构象多样性的袋里指标。结果显示,HF-S1 通常具有更高的标准差,说明它探索了更广泛的构象区域。尤其在蛋白质—抗体、蛋白质—RNA 和蛋白质—DNA 复合物中,HF-S1 生成了更多具有不同结构精度的候选构象,说明它能够引导采样进入多个可能的相互作用几何状态,而不是局限在单一局部区域。
研究团队还追踪了随着采样步数增加,模型累计生成构象的平均精度变化。HF3 的平均精度基本保持稳定,说明它的采样顺序近似随机;HF-S1 则在早期采样阶段精度较高,随后逐渐下降。这完全符合它的设计逻辑:模型优先使用高概率接触作为约束生成结构,随后逐步探索概率较低的接触配置。也就是说,HF-S1 不仅提升了采样效率,还能优先探索最有结构意义的构象区域。
两个代表性案例进一步说明了这一点。在 Gel4–Nb4 纳米抗体复合物和 Cas12m2–crRNA 蛋白质—RNA 异二聚体中,HF-S1 生成的构象覆盖了更宽的置信度和结构精度范围,并包含更多高精度预测;而 HF3 的预测集中在较窄区域,结构多样性和高质量候选都偏少。
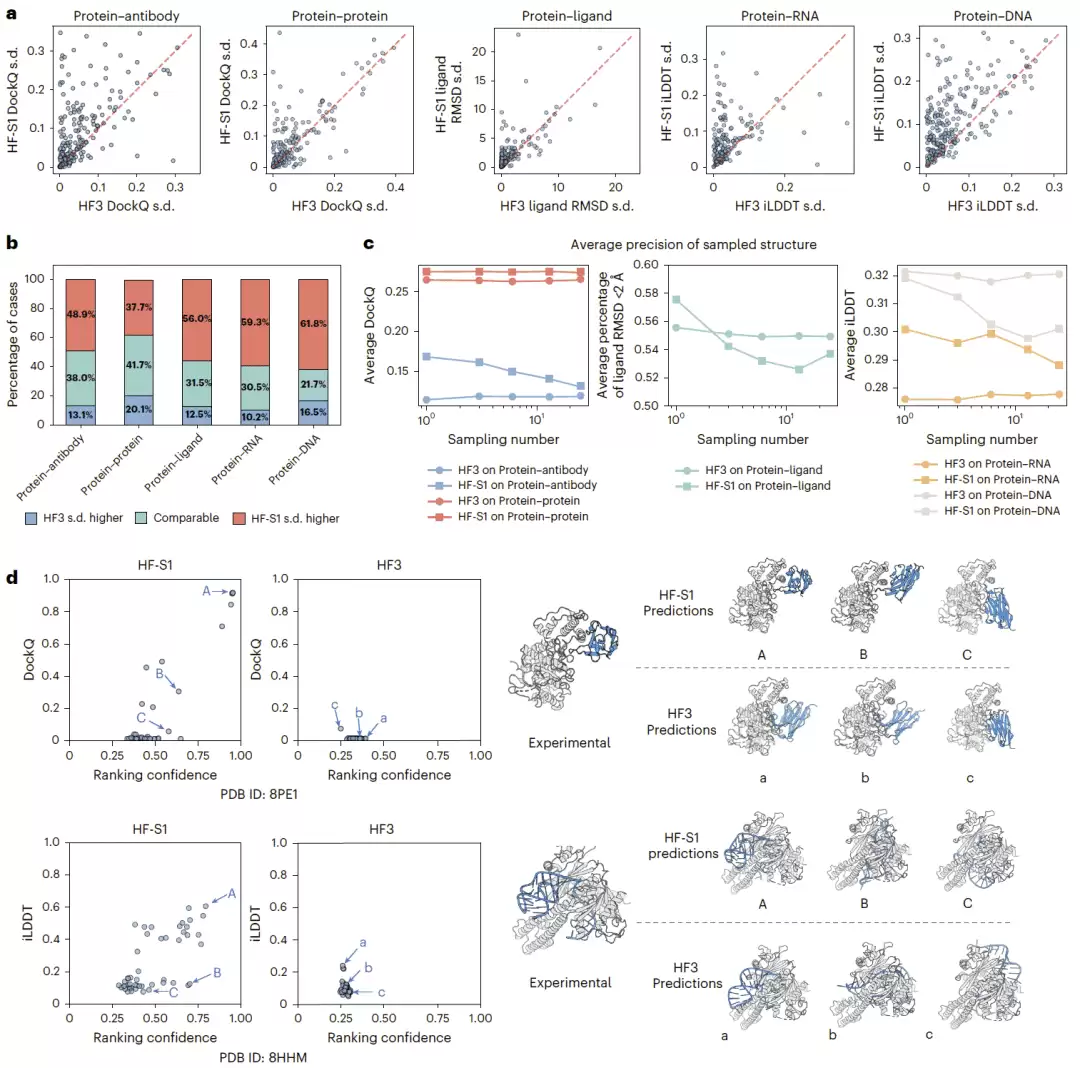
图4|引导式采样改善构象空间探索。
讨论
这项研究揭示了一个重要事实:大规模采样虽然是提高结构预测准确性的常规方法,但无引导采样效率太低,而且很难判断不同靶标到底需要多少采样。HF-S1 的核心贡献在于把结构预测中的构象搜索转化为一个更有规划的过程——先通过链间接触概率描绘构象空间蓝图,再把采样资源集中到最有信息量的相互作用区域。这样既提高了结构预测准确性,也显著降低了计算成本。
与传统“生成大量结构再筛选”的范式相比,HF-S1 的“搜索—筛选”策略更有助于跳出局部最优。在多表位抗体、隐蔽口袋或处于构象转换状态的受体等复杂系统中,生物学相关构象可能只占很小比例。引导式采样通过优先探索多个潜在结合模式,可以减少这些稀有但重要构象被遗漏的风险——其价值已经超越了单一结构预测本身。
不过必须承认,结构置信度评估仍然是限制因素。当前置信度评分有时会错误选择并非最准确的结构,高置信度结构不一定准确,而低置信度候选中也可能包含接近天然状态的构象。未来需要更精细的打分函数或基于集合的校准方法,才能更好地选择高质量候选结构。
此外,HF-S1 的引导式规划也可能出现收益递减。当前策略按照接触概率从高到低贪婪选择接触,后期可能逐渐采样到信息量较低的区域。未来可以引入基于接触饱和或结构质量平台期的提前停止机制,进一步提高效率。保持构象多样性同样重要——当前的冗余接触剪枝能减少重复采样,但过程偏串行,未来需要更主动、更高效的多样性保持策略。
总体来看,HF-S1 展示了一种更智能的构象空间导航方式。通过把链间接触概率作为结构生成蓝图,它能够在更少采样次数下生成更准确、更多样的生物分子复合物结构。如果未来能结合蒙特卡洛树搜索、强化学习等策略,在探索与利用之间动态平衡,复杂生物分子结构预测的稳健性和效率有望再上一个台阶。
参考资料
Liu, L., Liu, Y., Ye, X. et al. Reshaping biomolecular structure prediction through strategic conformational exploration with HelixFold-S1. Nat Mach Intell (2026).
https://doi.org/10.1038/s42256-026-01264-2




