单细胞扰动预测,通俗地说,就是让模型学会预测细胞在药物处理、基因编辑或其他干预手段下会发生怎样的状态变化。这是精准医疗与药物研发中的一个核心难题。然而,一个棘手的先天性挑战始终存在:单细胞测序具有破坏性,研究人员无法对同一个细胞同时测量其“用药前”和“用药后”的状态。因此,他们只能获得群体层面的分布数据,而非一一对应的配对样本。这使得传统的监督学习方法在这里难以施展,无法精准捕捉真实的扰动效应。更令人困扰的是,现有绝大多数模型必须针对每一种扰动单独训练,一旦遇到全新的药物、剂量或组合,模型基本就“没学过,无法应对”。
那么,有没有方法能让模型学会举一反三?近期的一项研究给出了一个巧妙的解决方案——Conditional Monge Gap(简称CMonge)。它基于条件最优传输(Optimal Transport)理论,通过引入条件信息来学习一个全局的最优传输映射。这样一来,不同药物、不同剂量、不同组合之间的知识可以共享,甚至还能推广到从未见过的全新条件。研究团队在单细胞RNA测序数据集SciPlex以及多重蛋白成像数据集4i上进行了系统验证,结果令人振奋:CMonge在已知扰动的预测任务上达到了甚至超越了当前最佳方法;而在预测从未见过的药物方面,它显著优于主流模型chemCPA,同时能够很好地保留细胞群体的异质性。更值得一提的是,如果引入药物结构信息并扩大训练规模,CMonge可以同时对数百种药物进行联合学习,并且仅凭药物的分子结构就能预测新药引发的响应——这为虚拟药物筛选和药物再利用打开了一扇新的大门。

理解细胞如何响应外界扰动,一直是生物医学研究的核心命题。单细胞RNA测序技术和高通量药物筛选的发展,使研究人员能够系统观察不同细胞群体对药物、基因编辑和各种治疗手段的反应。但问题在于,即使是最先进的实验平台,也无法穷举海量的药物结构、剂量组合和基因扰动空间。因此,利用机器学习来预测那些尚未被实验测量的扰动结果,成了一个非常实际的研究方向。
早期的方法有scGen,它用变分自编码器学习扰动前后的潜在空间变化;随后PerturbNet、GEARS、chemCPA乃至一些单细胞基础模型陆续登场。这些方法各有突破,但始终绕不开两个关键挑战。第一,由于单细胞测序会破坏细胞,扰动前后的细胞无法一一对应,模型必须学习分布之间的变化,而非样本之间的直接映射。第二,大多数模型缺乏对未见药物、新剂量、新扰动条件的泛化能力,说白了就是“没见过的就不会”。
最优传输理论在这个问题上天然具有优势——它能直接学习两个细胞群体分布之间的转换关系。之前的CellOT和scPRAM已经用最优传输建模过细胞状态变化,但它们都是“局部模型”:每一种药物或条件都要单独训练一个模型,既无法共享知识,也预测不了新条件。正是在这个背景下,CMonge被提出,目标就是用统一的条件最优传输框架来实现跨扰动学习和泛化预测。
方法
CMonge建立在Monge Gap最优传输框架之上。原来的Monge Gap是对每一种扰动单独学习一个最优传输映射,而CMonge引入了条件变量,将药物、剂量或药物组合信息编码成上下文嵌入,然后用一个统一模型同时学习多个扰动之间共享的传输规律。
模型的输入包括源细胞分布和条件嵌入信息。对于药物,研究者使用了两种表示方式:一种基于药物作用机制(Mode of Action,MoA)构建嵌入;另一种基于SMILES结构计算RDKit分子指纹。药物剂量则用对数剂量表示。模型通过多层感知机学习细胞状态的变化向量,然后加到原始细胞表示上,最终得到预测的扰动后状态。对于药物组合,还进一步采用了DeepSets结构来实现排列不变的组合表示。
评估是在SciPlex数据集(187种药物、4个剂量、约76万细胞)和4i蛋白成像数据集(35种癌症治疗方案)上进行的,分别测试了已知条件(In-Sample)和未见条件(Out-of-Sample)两类预测任务。
结果
CMonge建立统一的条件最优传输框架
先梳理一下整体设计思路。传统的最优传输方法需要为每一种扰动单独训练一个模型,不同任务之间完全无法共享知识。CMonge则用统一的条件模型同时学习多个扰动下的细胞状态转换规律,相当于构建了一个全局的最优传输估计器。这个设计有一个很直接的好处:推理时只要输入新的条件信息,模型就能直接预测对应的扰动后状态,完全不需要重新训练。研究团队认为,这种全局学习机制能够充分利用不同扰动之间潜在的生物学共性,从而显著提升泛化能力。
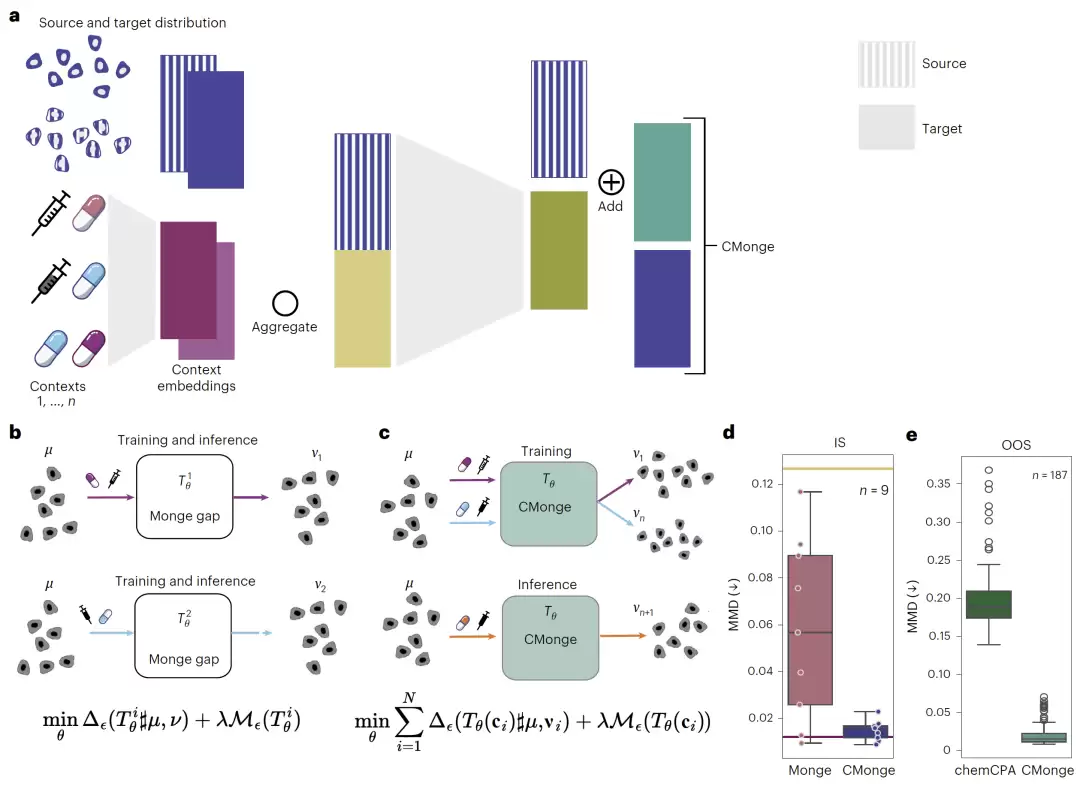
图1:Conditional Monge Gap总体框架示意图,包括条件编码、全局最优传输学习以及已知和未知扰动预测流程。
条件信息显著提升已知扰动预测能力
在SciPlex数据集上,研究团队首先验证了条件信息的重要性。实验发现,仅利用单独剂量信息构建的CMonge模型,就已经能恢复大部分性能损失,预测效果接近为每种药物单独训练36个Monge模型的表现。而一旦加入药物信息,性能还能进一步提升。尤其是在高剂量条件下,模型可以准确重建扰动引起的细胞状态变化。
有趣的是,当采用药物作用机制(MoA)作为条件嵌入时,单个CMonge模型就能达到甚至超过36个独立模型的整体表现。即使只使用药物结构指纹(RDKit),随着训练药物数量的增加,模型性能也在持续提升。
团队还专门分析了MEK抑制剂Trametinib的信号通路变化,发现CMonge预测得到的富集通路与真实实验高度一致,包括MAPK信号通路及其下游转录调控网络。这说明模型不仅能预测表达变化,还能保留重要的生物学机制。
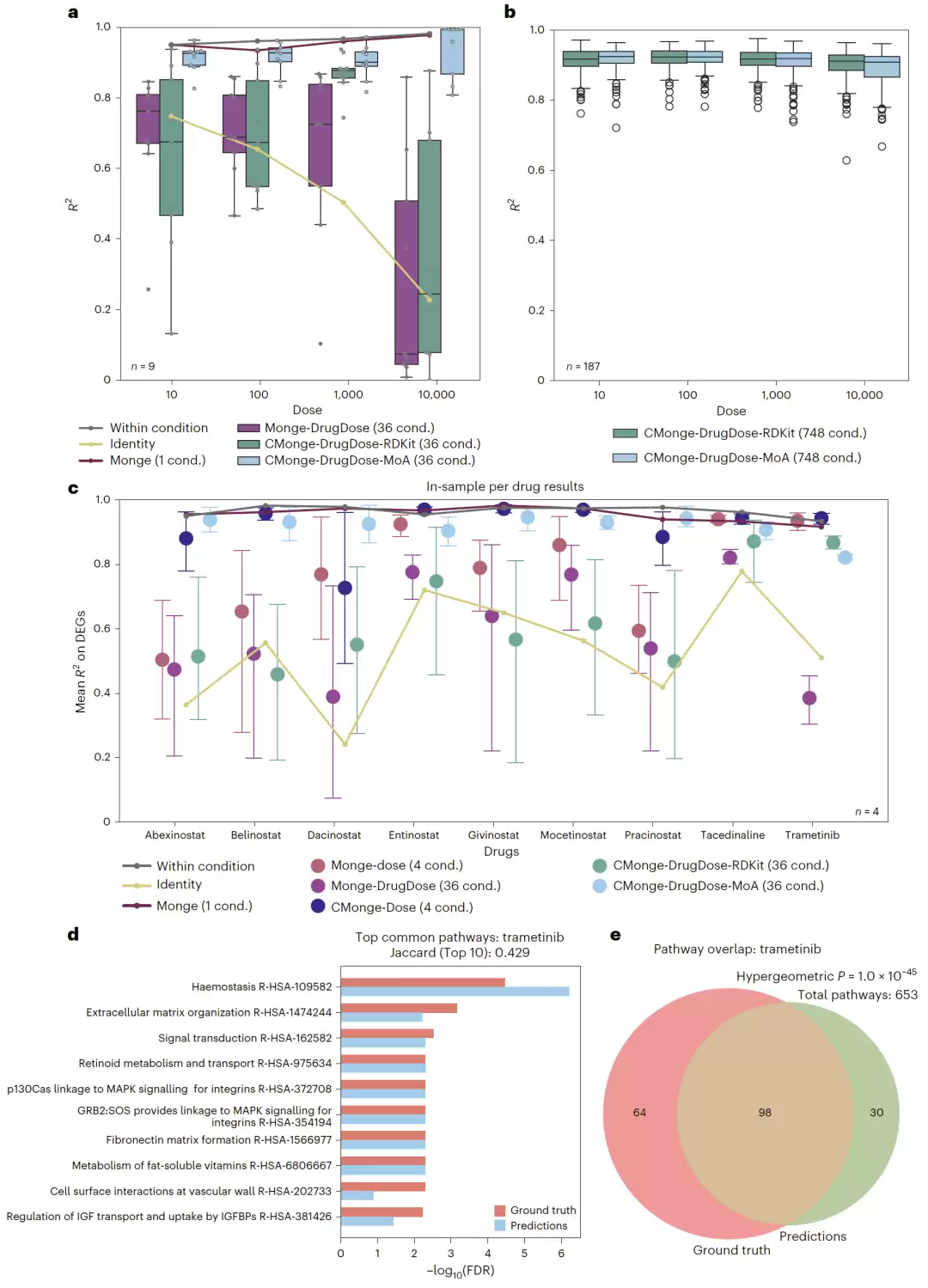
图2:SciPlex数据集中不同条件模型的性能比较,以及Trametinib通路富集分析结果。
扩展至数百种药物后结构信息发挥关键作用
在只包含少量药物时,基于MoA的条件嵌入明显优于基于RDKit结构特征的嵌入。这是否是因为训练规模不够?为了验证这一点,团队将训练数据扩展到了SciPlex的全部187种药物,一共748个药物-剂量组合。结果,RDKit模型的性能显著提升,并达到了与MoA模型相近的水平。
这个结果说明,随着药物数量增加,模型能够逐渐学会从分子结构中提取与生物学效应相关的信息。由于RDKit嵌入只依赖药物结构,不需要任何实验测量数据,这为预测全新药物的响应提供了非常重要的基础。
另外值得注意的一点是,CMonge的计算复杂度近似随着条件数量线性增长,相比为每种药物单独训练模型,效率优势非常明显。
实现未见药物与未见剂量预测
真正考验泛化能力的,是模型在从未见过的条件下表现如何。对于未见剂量预测,传统的无条件模型性能会迅速下降,但加入剂量信息后的CMonge能够显著提高预测精度,即使是面对训练过程中从未出现过的剂量水平,依然能保持良好表现。
更具挑战性的是未见药物预测。团队采用“留一药物”策略——把某一种药物的所有剂量条件全部从训练集中移除。结果显示,基于MoA嵌入的CMonge几乎达到了条件特异模型的理论上限,远超当前主流方法chemCPA。
在进一步扩大到187种药物的交叉验证实验中,基于RDKit结构表示的CMonge同样超过了chemCPA。无论用R²、Wasserstein距离还是MMD来评价,CMonge都是最佳。尤其在高剂量条件下,优势更加明显。通过UMAP可视化可以看到,CMonge预测得到的细胞群体不仅准确落在真实目标分布附近,而且能够很好地保持细胞群体内部的异质性结构;相比之下,chemCPA往往只能学习平均效应,预测结果出现了明显的模式坍塌。
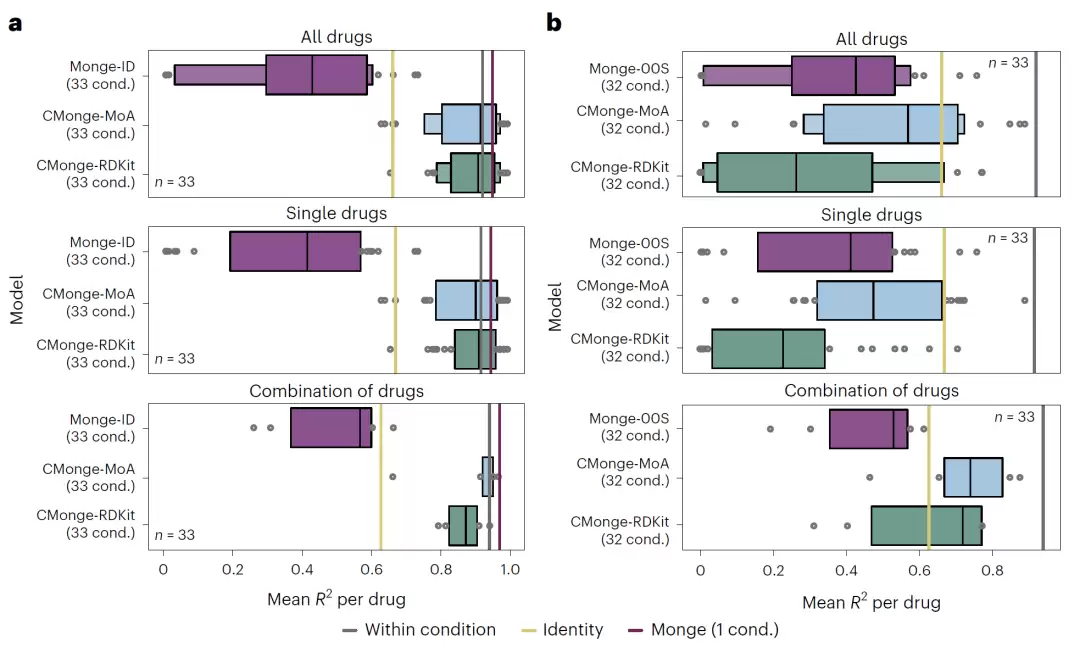
图3:4i数据集与SciPlex数据集中的未见条件预测结果比较。
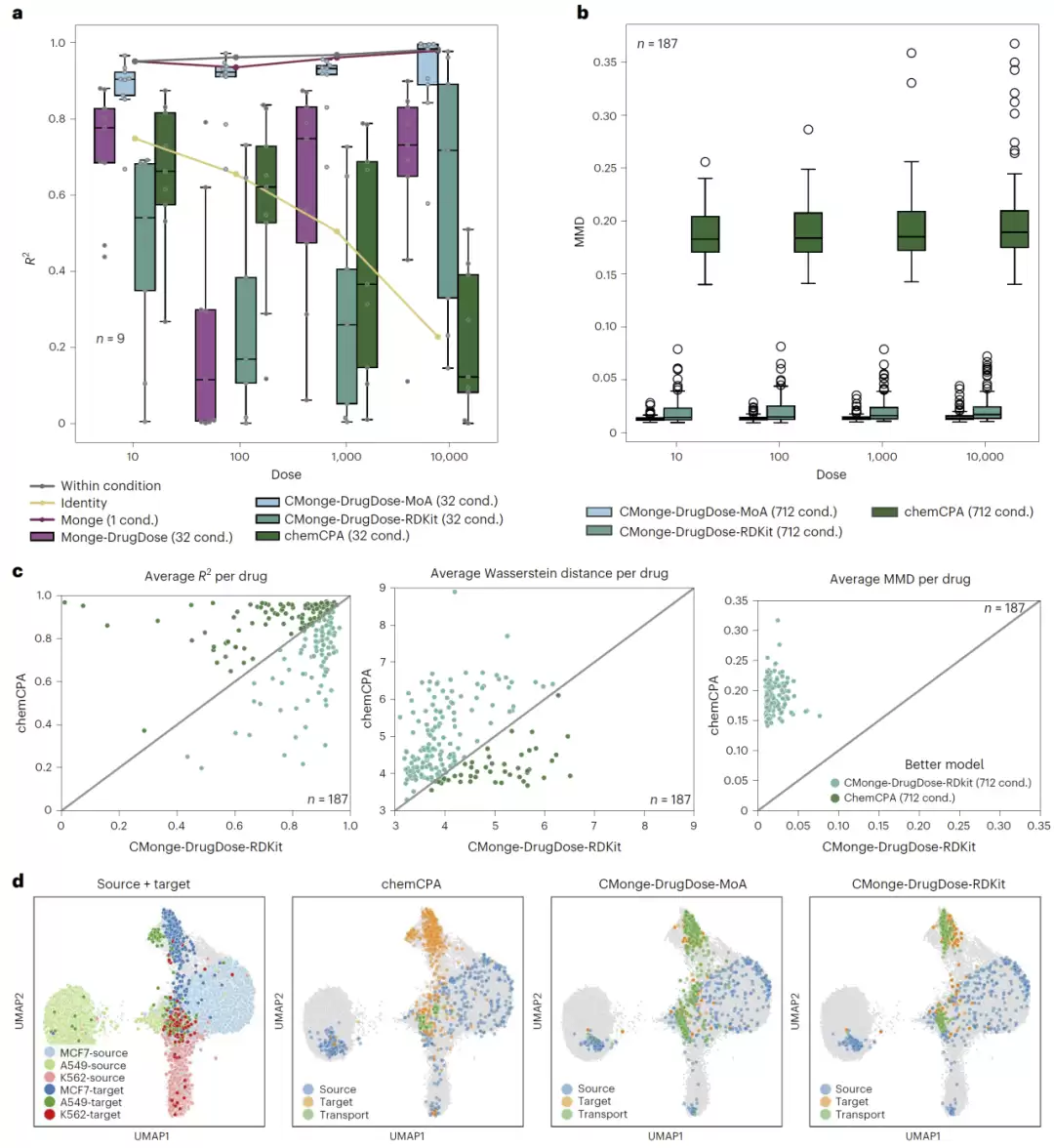
图4:CMonge与chemCPA在未见药物预测中的性能比较及UMAP可视化结果。
讨论
Conditional Monge Gap为单细胞扰动建模提供了一套全新的条件最优传输框架。与传统方法相比,CMonge不再需要为每种扰动单独训练模型,而是通过统一模型实现跨任务学习和知识共享,泛化能力得到了显著提升。
研究结果表明,CMonge不仅在已知扰动预测中达到了当前最佳水平,而且在未见药物和未见剂量预测任务上也表现出色。特别是利用药物结构信息时,模型能够直接从分子结构推断潜在的细胞响应,这为虚拟药物筛选和药物再利用提供了一条切实可行的技术路径。
有意思的是,相比近年来兴起的单细胞基础模型,CMonge的参数量极小,却在多个基准测试中取得了更好的结果。这或许说明,针对具体科学问题设计的物理和数学归纳偏置,可能比单纯扩大模型规模更有效。
当然,当前模型仍有局限。比如,对于训练数据中极少出现的药物类别、高剂量条件以及完全未知的细胞类型,泛化能力还有提升空间。未来如果结合更复杂的注意力机制、流匹配方法以及更大规模的单细胞筛选数据集,预测精度有望进一步提高。
总体来看,CMonge展示了条件最优传输在单细胞生物学中的巨大潜力。通过把药物结构、剂量和组合信息统一纳入建模框架,研究团队为构建真正可泛化的“虚拟细胞模型”迈出了关键一步,也为精准医疗和智能药物发现提供了新的技术路线。
参考资料
Driessen, A., Rajwade, D.A., Harsanyi, B. et al. Conditional Monge Gap enables generalizable single-cell perturbation modelling. Nat Mach Intell (2026).
https://doi.org/10.1038/s42256-026-01242-8