你是否曾深思,当深度学习模型将DNA序列仅仅视作一串由A、T、C、G组成的文本字符时,它究竟遗漏了哪些关键信息?
诚然,仅着眼于碱基字母的排列顺序,现有模型已能发掘出不少调控线索。然而现实中的DNA绝非单行文本,它是一条由两条方向相反、彼此互补且动态协同的双链分子构成。这种固有结构引出一个不容回避的问题:基础模型能否更进一步,不只是“读取序列”,而是真正领悟正链与反向互补链之间存在的内在关联?

近期,来自湖南大学的曾湘祥教授团队,联合隆平农学院于峰教授、生物学院罗宵教授等,在权威期刊《自然·机器智能》(Nature Machine Intelligence)上发表了关于CrossDNA的研究成果,论文标题为Explicit Dynamic Cross-Strand Interactions for DNA Sequence Language Modeling。该研究聚焦于DNA双链间的信息交互机制,设计了一套显式、动态的序列建模框架——其核心理念非常直接:让语言模型更贴近DNA分子原本的结构与功能逻辑。
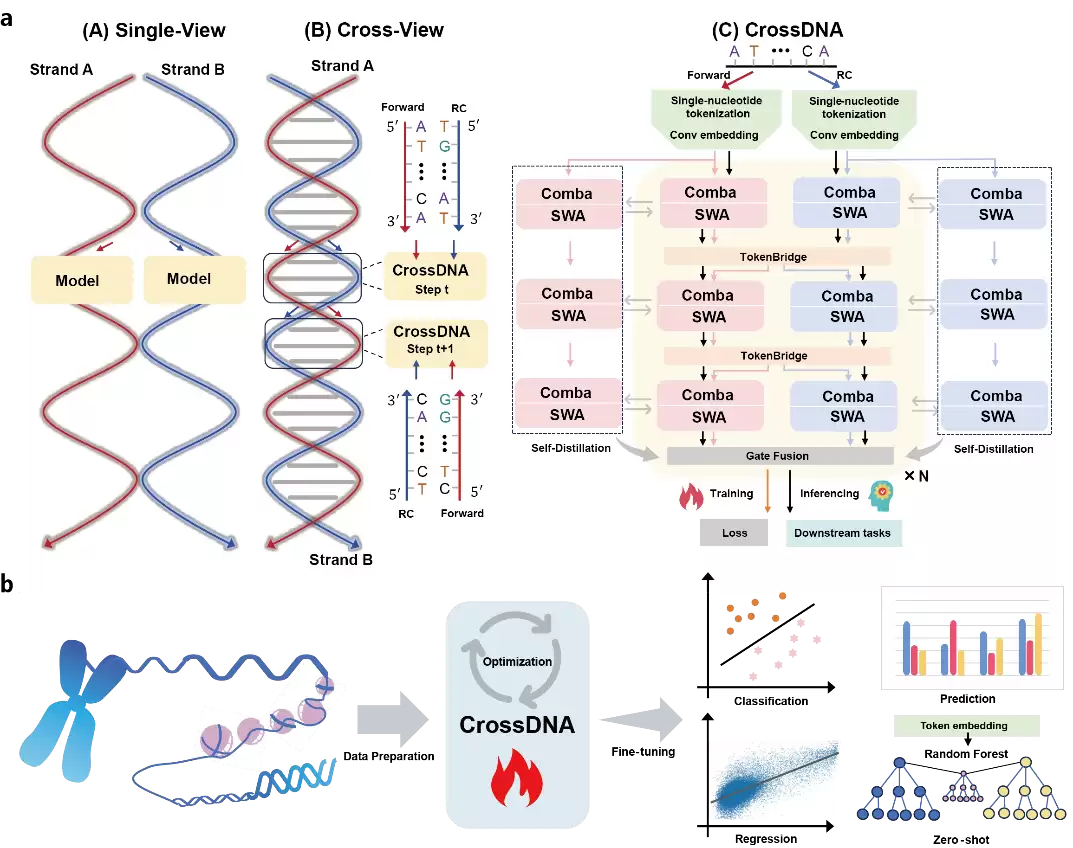
图 1: CrossDNA整体工作流与双分支架构示意图。
从单链序列走向双链动态交互
DNA序列语言建模的目标在于,通过学习基因组序列的表示方法,进而服务于功能元件注释、调控机制解析、非编码变异解释以及长程基因组预测等关键任务。当前大多数方法沿袭单链建模路线——将DNA视为单向或双向文本,再借助反向互补数据增强、参数共享或等变结构等静态约束,使模型获得一定程度的方向一致性与通用表征能力。
但根本问题在于,真实生物系统中的DNA从来不是孤立存在的单链。两条链之间不仅存在物理耦合,还在转录调控、功能元件识别及变异效应传递中展现出深层次的功能协同。相较于单链建模,双链建模强调在序列学习过程中,模型应显式地把握正链与反向互补链之间的上下文交互与动态信息传递——这对于理解基因组结构与功能的关系具有重大意义。
CrossDNA正是瞄准了这一痛点:它希望模型在表示学习阶段就直接理解两条链之间的协同关系,而不是等到输入前或输出后才去补全这一关键信息。
Cross‑View、TokenBridge 与自蒸馏机制
CrossDNA采用了一种双分支语言模型架构。两个分支结构相同但参数不共享,各自处理同一基因组区域的正向链和反向互补链。训练过程中,一个名为Cross-View的机制会将双链相邻片段交替送入两个分支——想象一下,模型在训练时不停切换观察角度,而不只盯着DNA的一个面。
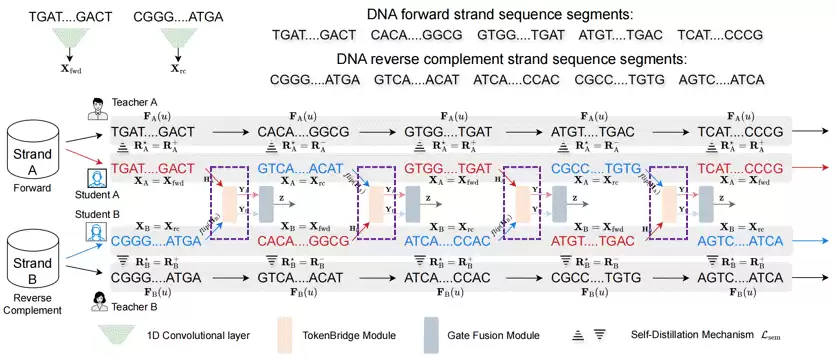
图 2: Cross-View下的数据预训练过程。
在每个分支内部,CrossDNA使用Comba-SWA结构来建模局部模式与长程依赖。随后,模型将两条链的特征对齐到统一的正向坐标系,再通过一个轻量级TokenBridge模块,在碱基标记层面完成跨链信息交换。最后利用门控融合模块,将两条链的信息整合成下游任务可用的表示。
为防止交替输入造成表示的不连续性,CrossDNA还引入了基于指数滑动平均教师模型的自蒸馏机制——为两个学生分支施加语义一致性约束。目的十分明确:让模型在不同链方向之间维持稳定的表示,而不是仅仅记住某个输入方向。
基准任务:调控元件分类表现优异
论文首先在基因组基准测试(Genomic Benchmark)的八项调控元件分类任务上进行了评估。在参数规模匹配的前提下,408K参数版本的CrossDNA取得了平均准确率88.2%,并在小鼠增强子、编码区与基因间区识别、人类调控序列、人类开放染色质区域、人类非TATA启动子这五项任务中排名第一。
这一结果的意义在于:CrossDNA并非依靠堆砌参数取胜。在紧凑的参数量下,显式双链建模带来的性能提升是实实在在的。论文同时指出,在同一组任务上,它的平均准确率甚至高于某些参数规模更大的模型。这一点值得深思——对DNA序列建模而言,模型能否理解生物结构先验,其重要性或许不亚于模型本身的规模大小。
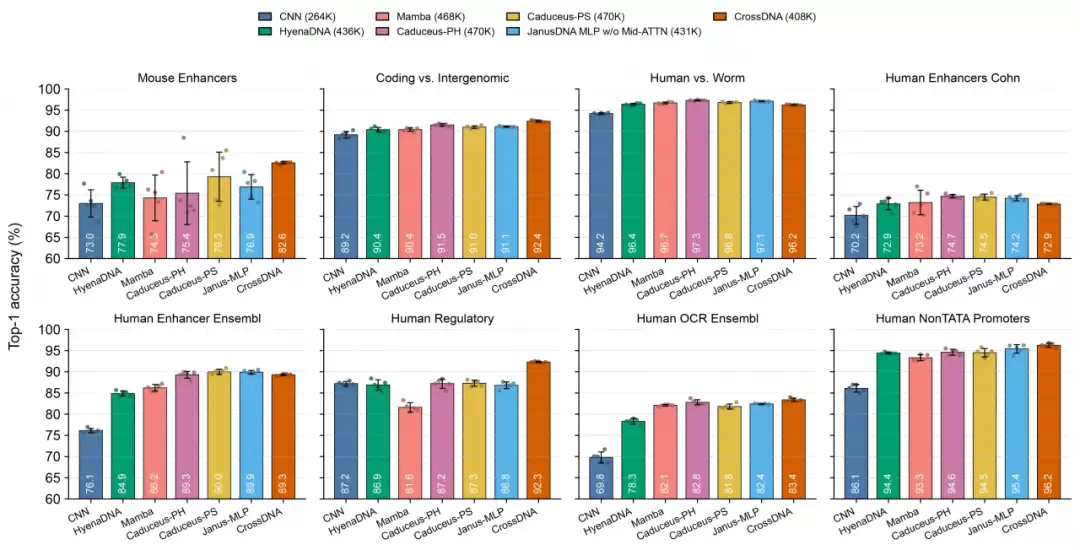
图 3: 不同DNA语言模型在小参数上的架构性能优势比较。
在Nucleotide Transformer基准上的评估
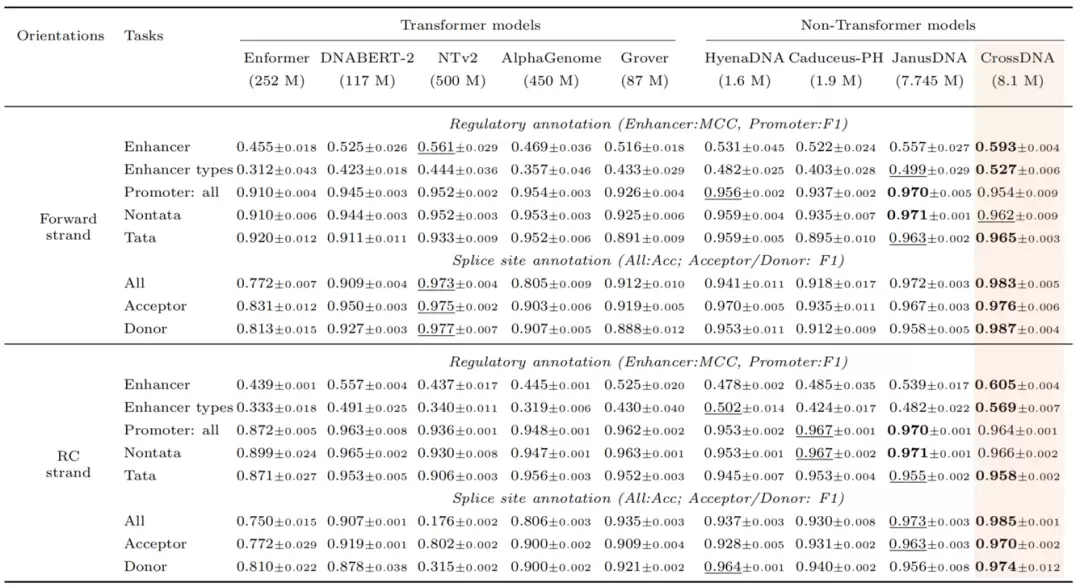
对于DNA模型来说,方向一致性是一个基础却关键的问题。同一段DNA与其反向互补序列指向相同的基因组位置——如果模型在两种方向上给出截然不同的判断,那么全局扫描、功能元件识别以及变异预测的可靠性都会大打折扣。
论文在Nucleotide Transformer任务集中分别测试了正向链和反向互补链的表现。CrossDNA在全部36个相关下游任务中有33项排名第一或第二,并且在增强子、启动子、剪接位点及组蛋白标记等任务上,方向一致性保持得相当出色。据论文报告,它在代表性任务上的正反链分数差异通常低于0.015,最大值不超过0.042——这为“显式跨链建模有助于降低方向性预测波动”这一判断提供了有力支撑。
表 1: 不同DNA基础模型在Nucleotide Transformer benchmark上的性能比较。
预测、泛化性能与长距离任务测评
在人类K562细胞系、小鼠和果蝇的增强子功能元件独立数据集上,CrossDNA的泛化性能展现出明显竞争力(图4 b,c,d,e,g,h)。在难度较高的小鼠数据集上(图4 g,h),经过小鼠基因组上的持续预训练后,它依然保持了领先优势。此外,对于新增强子功能序列元件的发现,CrossDNA给出了置信度很高的预测分数(图4 f)。
长程任务方面,CrossDNA在eQTL任务中达到了先进或持平的预测性能(图4 i)。而在增强子-靶基因交互预测中,Cross-View机制提供的双链视角上下文信息,使得交叉验证时正样本的预测得分高于单链视角——这直观地表明:双链信息确实带来了额外的帮助。
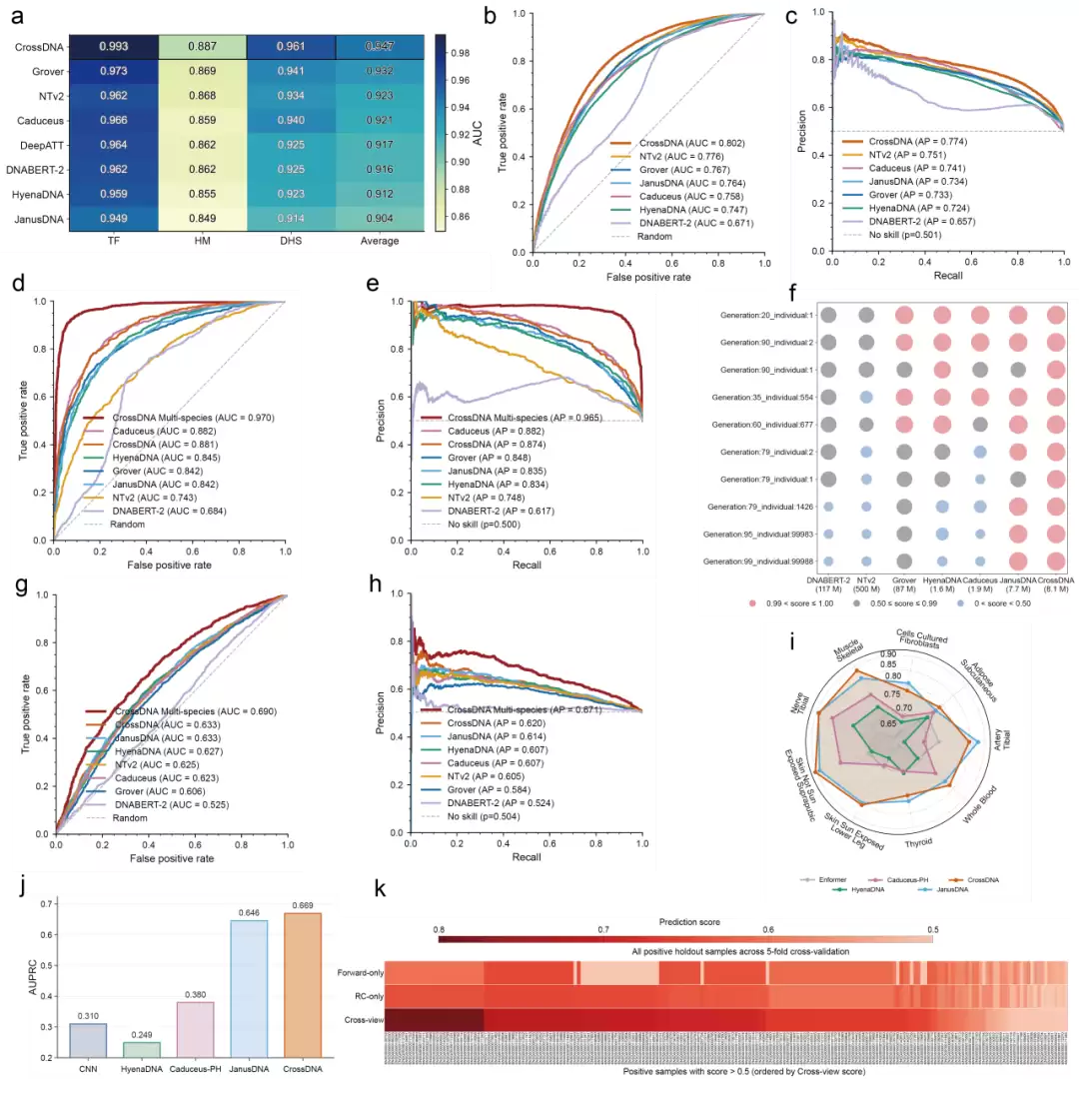
图 4: CrossDNA在泛化任务、预测任务、长程任务上的表现。
表征质量:零样本嵌入评估
除了监督微调,论文还评估了CrossDNA作为通用序列表征模型的能力。研究者从42个真实世界序列分类任务中提取了零样本嵌入表示,再用随机森林分类器进行测试。结果显示,CrossDNA相对于JanusDNA、DNABERT-2、Grover、NTv2、Caduceus-PH和HyenaDNA等模型,均取得了正向差异。相比JanusDNA的提升幅度虽然不大但保持稳定,而相对于HyenaDNA和Caduceus-PH,差距则更为明显。
这意味着,CrossDNA在序列级别的特征嵌入上确实展现出较高的特征质量,能够支撑可靠的功能元件分类分析。
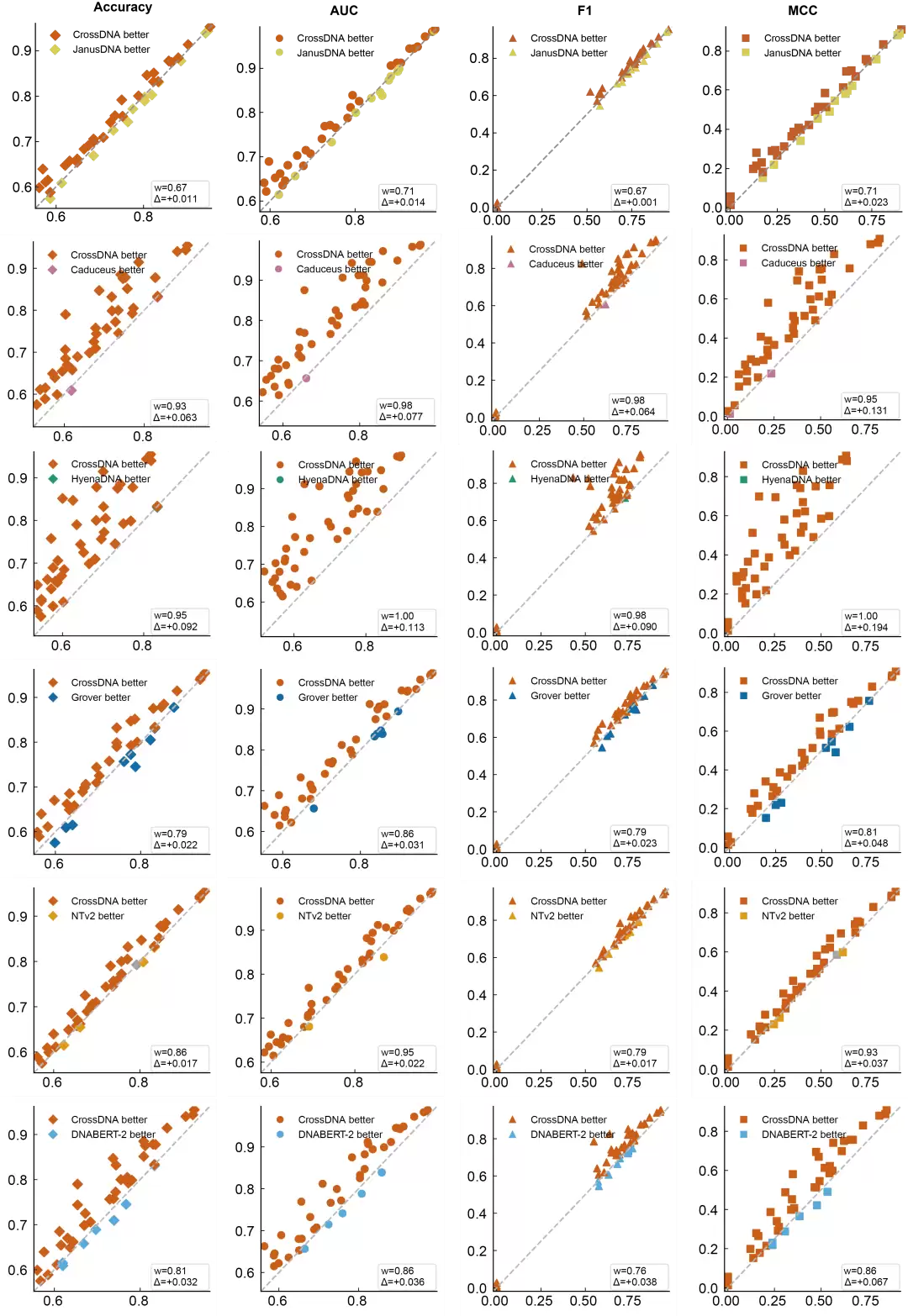
图 5: 零样本嵌入性能比较。
生物解释:motif、非编码变异与候选增强子
一个优秀的DNA基础模型,仅仅给出分数是不够的——它还需要帮助研究者理解哪些序列片段在发挥关键作用。论文从生物解释的角度做了进一步分析。
通过计算机模拟突变分析,模型识别出的高贡献区域能够对应到已知的转录因子结合基序——例如DREAM合成增强子中的NKX2-8和FOSL1信号(图6 a)。这说明CrossDNA的预测并非依赖表面序列偏差,而是真正捕捉到了与调控功能相关的序列模式。
在疾病相关变异分析中,论文重点讨论了冠心病相关变异rs113716316。CrossDNA将其优先定位到一个潜在的心脏增强子,并关联到FGR基因调控(图6 c,d,e,f)。模型结果提示,该变异可能涉及RUNX相关抑制信号减弱与AP-1相关活化信号增强——这为非编码变异如何影响疾病风险提供了一个具体的调控解释。
研究团队还利用CrossDNA扫描了K562细胞中未注释的区域,识别出748个高置信度的候选增强子。这些区域富集了SMAD3、TAL1/SCL、ERG等造血调控相关基序,在从头基序发现分析中还恢复了MYB家族和E2F家族等已知调控模式(图6 g,h)。

图 6: CrossDNA可解释性分析。
局限与未来展望
论文也坦诚地指出了CrossDNA当前存在的提升空间。早期预训练主要基于人类参考基因组,对个体水平的基因表达差异预测能力仍有限。未来若能引入群体遗传变异、多物种基因组以及更丰富的调控背景信息,模型对复杂调控现象的刻画能力预计还能再上一个台阶。
总体来看,CrossDNA的意义不仅限于性能提升。它提出了一种更贴近DNA分子结构的建模思路——将基础模型从单链序列建模推进到显式、动态的双链交互建模。它并非简单地把模型做得更大,而是将一个生物学事实重新置于模型中心:DNA的信息不仅来源于双链结构,更源自两条链之间的对应、互补与约束关系。
从这个角度而言,CrossDNA不只是一个新模型,更像是一个信号:下一代基因组基础模型,可能不全靠规模说了算,而是要更认真地把生命分子的结构规律融入语言模型之中。
参考资料
Yang, C., Liu, Y., Ling, L. et al. Explicit Dynamic Cross-Strand Interactions for DNA Sequence Language Modeling. Nat Mach Intell (2026).
https://www.nature.com/articles/s42256-026-01249-1




