AI药物发现中的核心悖论:我们最想要的,恰恰是模型最不擅长的
机器学习正以前所未有的速度重塑早期药物发现,这已成为行业共识。但在这一进程中,存在一个根本性的矛盾——真正有价值的发现,往往是模型最畏惧的挑战。
药物研发追求的是结构新颖的活性分子(hits),它们需要填补未被满足的临床需求、绕过现有专利壁垒、克服已有的耐药性问题。换言之,那些具有潜力的发现,通常位于已知化学空间之外。然而,机器学习模型在面对训练数据之外的分子——即常说的OOD问题——时频繁出现失误。这一问题在分子层面尤为突出:分子属于离散数据,仅改动一个原子或一个环系,就可能使分子脱离模型熟悉的分布范围。
更令人棘手的是数据稀缺问题。高质量的活性标注实验既昂贵又耗时,训练集往往只有几百个分子;而真正需要筛选的化合物库,动辄数十亿个。一边是几百个“见过”的分子,另一边是数十亿个“闻所未闻”的分子,这种巨大的分布漂移,使得利用机器学习发现结构新颖的先导化合物变成了一项近乎西西弗斯式的任务。
于是核心问题浮现:当模型步入它所学化学空间的边缘之外,我们凭什么相信它的预测?
最近一篇论文给出了一个相当优雅的解决方案——一个被称为“不熟悉度”(unfamiliarity)的新指标。
过去的解法各有盲区
在介绍新方法之前,有必要回顾过去人们走过的两条路径。
第一条是适用域(applicability domain)思路。简单来说,就是在化学空间中给训练数据划定一个范围,范围内的分子被认为可以可靠预测,范围外的即为OOD。这个范围通常通过训练分子的相似度阈值来定义。问题是,这种方法完全没有利用模型自身学到的信息,而且本质上是基于相似度,反而会将结构新颖的分子悉数排除——这恰恰与寻找新分子的目标背道而驰。
第二条是不确定性估计(uncertainty estimation),利用模型自身的预测置信度,通常通过概率建模实现。这条路径理论上允许考虑结构新颖的分子,但存在一个致命弱点:面对OOD样本时,它常常给出过度自信的预测。模型可能信誓旦旦地告诉你某个它从未真正理解的分子具有活性,而你却无从分辨。
两条路径各有盲区,而这篇论文要补上的,正是中间那块拼图。
核心洞见:会复述,才是真懂
论文的关键想法,藏在一个看似简单的类比里:一个真正理解某样东西的人,能够凭记忆将其准确地复述或重画出来;而对于一知半解的东西,复述往往支离破碎。
把这个直觉迁移到分子上:如果一个模型能将某个分子压缩进自己的内部表示,再解压还原成原来的样子,说明这个分子对模型来说是熟悉的;反之,如果还原得乱七八糟,说明这个分子落在了模型学过的分布之外。
基于这一假设,作者提出用重建能力(reconstruction ability)作为衡量分子是否OOD的直接指标,并由此定义了不熟悉度。这实际上是对传统做法的反转。在生成式化学中,自编码器的重建任务通常是为了生成新分子,或者提升预测性能;而这篇论文将重建质量本身作为OOD检测的信号。
方法:联合分子模型
具体如何实现?作者搭建了一个称为联合分子模型(Joint Molecular Model,JMM)的架构。它本质上是一个半监督的自编码器,可以拆分为三个部分:
编码器:分子先被表示为SMILES字符串(一种用文本编码分子拓扑与原子、键类型的方式),然后一个一维卷积神经网络(1D-CNN)将SMILES编码成一个压缩的潜在向量z。
解码器:一个带长短期记忆(LSTM)的循环神经网络,将潜在向量z再解码回SMILES字符串。
预测头:同样的潜在向量z,被送入一个近似贝叶斯分类器,用于预测分子性质(例如生物活性),同时估计预测的不确定性。
关键在于“联合”二字:重建任务和性质预测任务共享同一个潜在表示z,并且一起训练。这样既保证了潜在空间同时为两个任务编码了相关信息,又让重建质量能够反映模型对这个分子的理解程度。
整个编码器-解码器先在ChEMBL的约120万个无标注分子上进行了预训练,让模型先学会SMILES字符串的语法,然后利用每个带标注的数据集进行微调。
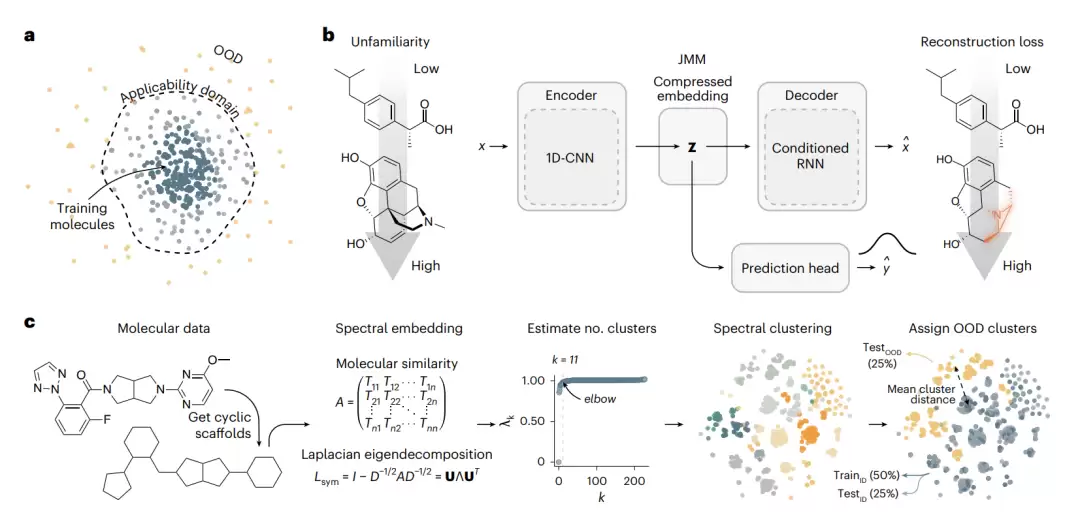 图1 | 用联合建模估计分子数据的不熟悉度。a,适用域概念示意:在化学空间中靠近训练数据的分子位于模型适用域内,边界之外的分子被视为OOD。b,JMM的架构:通过重建损失估计一个分子对模型有多不熟悉。c,通过谱聚类将分子数据分成同分布组与OOD组,从而制造分子分布漂移(以OX2R数据集为例)。
图1 | 用联合建模估计分子数据的不熟悉度。a,适用域概念示意:在化学空间中靠近训练数据的分子位于模型适用域内,边界之外的分子被视为OOD。b,JMM的架构:通过重建损失估计一个分子对模型有多不熟悉。c,通过谱聚类将分子数据分成同分布组与OOD组,从而制造分子分布漂移(以OX2R数据集为例)。
不熟悉度的数学定义其实很直接。重建损失被定义为所有SMILES token的负对数似然(按token长度归一化),而不熟悉度就是重建损失取对数。重建损失越低,不熟悉度越低,反之亦然。一句话:模型将这个分子还原得越糟糕,它对这个分子就越不熟悉。
验证:检测分布漂移
光有想法当然不够,还得看它是否真的有效。
作者收集了33个带实验标注的数据集,覆盖多种生物学性质和规模。为了制造可控的分布漂移,他们采用了一个巧妙的方法:先对分子的环骨架(去掉环外取代基的核心环系)进行谱聚类,确保结构相似的分子被聚在一起;然后将彼此距离最远的那些簇(约占25%的分子)拿出来作为OOD测试集(testOOD),剩下的分成训练集(trainID,约50%)和同分布测试集(testID,约25%)。
第一个问题:这样划分出来的testOOD,真的与其他两组来自不同分布吗?作者使用三种互补的相似度度量(ECFP指纹相似度、结构核心重叠MCS、CATS药效团相似度)进行验证,结果一致:testOOD与训练集、testID之间均存在统计显著的差异,而训练集和testID之间没有差异。分布漂移制造成功。
第二个问题:这种漂移真的会损害预测性能吗?确实如此。作者训练了几个成熟的基线模型(随机森林加ECFP、多层感知机加ECFP、随机森林加CATS),它们在testOOD上的表现一致地差于testID。这正是OOD数据的典型特征。
真正的考验:作者在每个数据集上计算出所有分子的不熟悉度,观察testOOD和testID是否有区别。结果非常清晰:testOOD分子的不熟悉度显著高于testID分子。
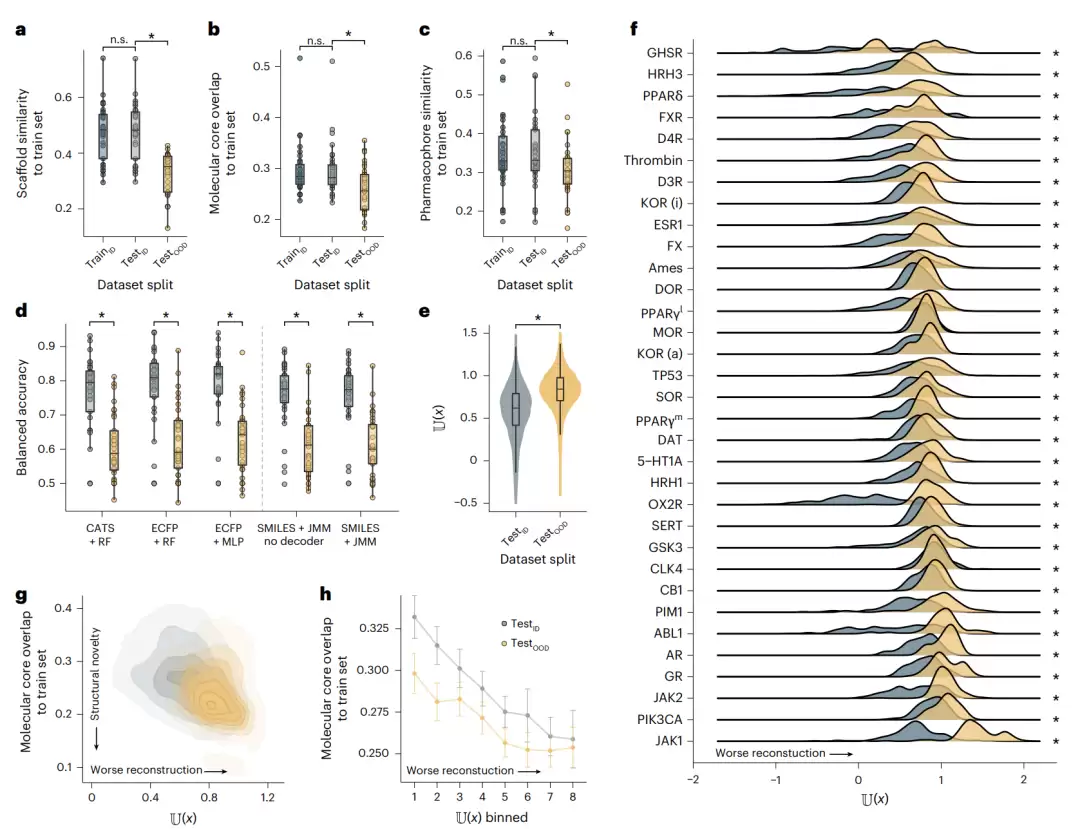 图2 | 用不熟悉度检测人为制造的分子分布漂移。a–c,三种相似度度量下各数据划分相对训练集的相似度。d,各基线模型与JMM在测试集上的预测性能。e,所有数据集中testID与testOOD分子的不熟悉度分布。f,逐数据集的不熟悉度分布。g–h,不熟悉度与分子核心重叠相似度的关系。
图2 | 用不熟悉度检测人为制造的分子分布漂移。a–c,三种相似度度量下各数据划分相对训练集的相似度。d,各基线模型与JMM在测试集上的预测性能。e,所有数据集中testID与testOOD分子的不熟悉度分布。f,逐数据集的不熟悉度分布。g–h,不熟悉度与分子核心重叠相似度的关系。
更重要的是一个排除性的发现:这种不熟悉度的差异,并不是由SMILES字符串长度、复杂度、分支数、分子图复杂度、分子量或官能团数量驱动的。换句话说,模型重建分子的能力,取决于这个分子离训练分布有多近,而不是这个分子本身有多复杂。这正是不熟悉度作为OOD指标的合理性所在。
顺带提一下性能问题:JMM在testID上的平衡准确率为0.75,略低于纯ECFP模型的0.78,但差距很小,与现有文献中SMILES模型与ECFP模型的对比一致。而且关键是,是否添加重建解码器对分类性能没有影响。也就是说,解码器让我们几乎免费获得了不熟悉度估计,而没有牺牲分类器的性能。
和其他指标比,强在哪?
不熟悉度好用吗?作者将其与三类成熟的可靠性度量进行了系统对比:数据驱动的训练集相似度、模型驱动的嵌入距离(分子z向量到训练集嵌入的马氏距离)、以及模型驱动的预测不确定性。
几个关键结论值得拿出来分享。
第一,所有这些指标都与模型性能相关,但不确定性和不熟悉度的相关性最强。当不确定性或不熟悉度较高时,模型的活性预测错误更多;反之则预测更准。
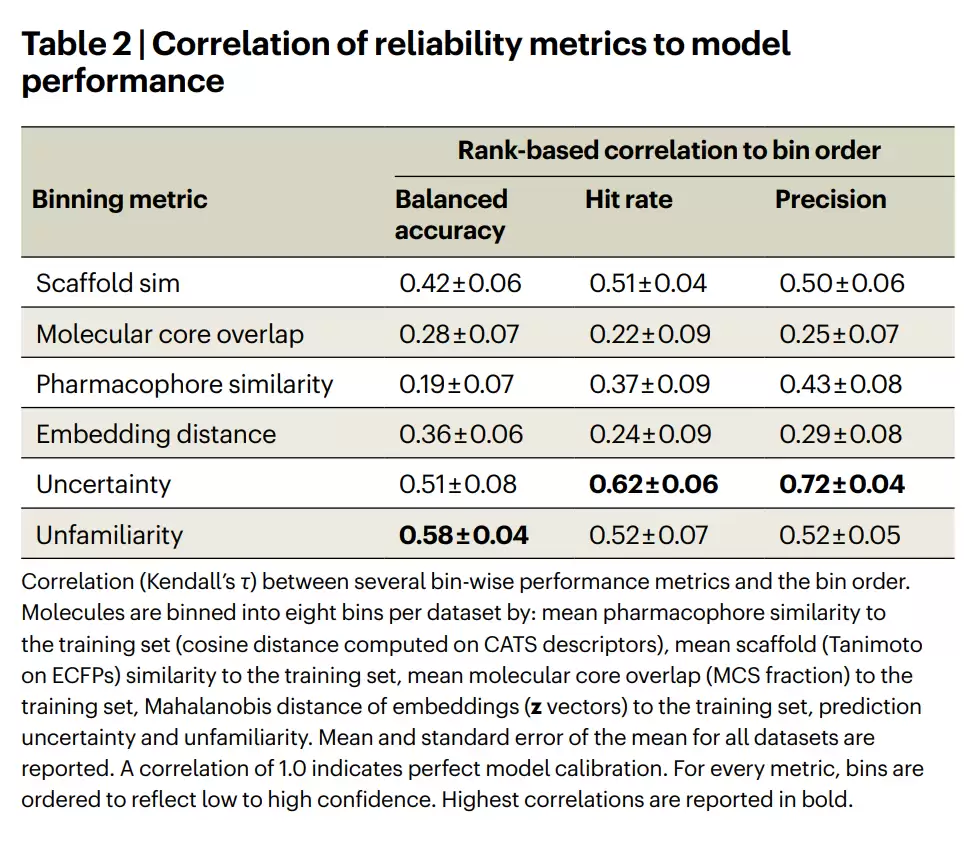 表2 | 各可靠性指标与模型性能的相关性。各指标(用于分箱)与平衡准确率、命中率、精确率之间的秩相关(Kendall's τ)。相关系数为1.0表示完美校准。最高相关性以粗体标出。
表2 | 各可靠性指标与模型性能的相关性。各指标(用于分箱)与平衡准确率、命中率、精确率之间的秩相关(Kendall's τ)。相关系数为1.0表示完美校准。最高相关性以粗体标出。
第二,也是最有意思的一点:不熟悉度和不确定性这两个指标本身几乎不相关(Spearman相关系数仅0.10)。这意味着它们捕捉的是关于预测可靠性的互补信息——这正是引入不熟悉度作为不确定性补充的理由。两个指标可以并用,从两个不同角度判断同一个预测是否可靠。
第三,模型驱动的指标全面优于数据驱动的指标。这说明模型能够从训练数据中提取出预定义的相似度度量无法捕捉的信息。
第四,不熟悉度甚至比嵌入距离更好。这有点反直觉——嵌入距离使用的是分类器直接使用的内部分子表示,按理说应该很有信息量。但结果是:一个模型能否从内部表示重建出分子,比这个内部表示本身更能反映预测的可靠性。简单的嵌入距离度量,无法捕捉那些影响具体任务结果的化学细微差别。
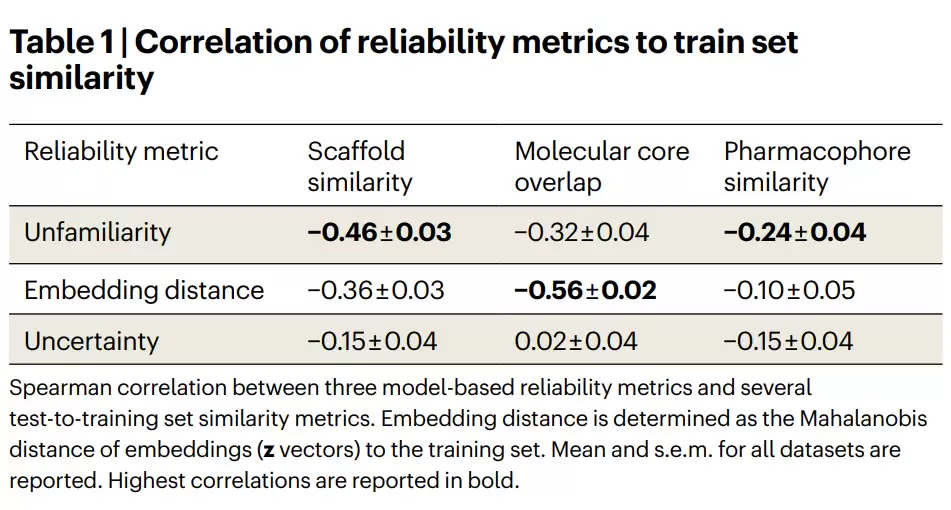 表1 | 三种模型驱动的可靠性指标与训练集相似度的相关性。不熟悉度、嵌入距离、不确定性分别与三类测试集-训练集相似度之间的Spearman相关系数。最高相关性以粗体标出。
表1 | 三种模型驱动的可靠性指标与训练集相似度的相关性。不熟悉度、嵌入距离、不确定性分别与三类测试集-训练集相似度之间的Spearman相关系数。最高相关性以粗体标出。
作者用一个漂亮的概率视角总结了三者的区别:嵌入距离主要反映p(x)(分子本身的分布),不确定性关联p(y|x)(给定分子的标签分布),而不熟悉度整合了两者,提供了对p(y, x|x)的洞察。这使得不熟悉度成为一个将结构信息与预测置信度联系起来的整体性可靠性指标。
放到真实的大规模筛选里,差异更戏剧
小数据集上的结论,在真实的大规模筛选场景中是否依然成立?
作者将分析扩展到了140万个分子(来自Asinex、Specs、Enamine三个商业筛选库)。这些库虽然没有活性标注,无法直接评估性能,但分子数量更大、更多样,更接近真实的虚拟筛选场景。
结果揭示了不熟悉度和不确定性之间一个相当戏剧性的差异。
筛选库的分子与训练集的结构重叠,比testOOD还要低。按照之前的结论,这意味着我们应该预期性能会进一步下降。
但是,预测不确定性几乎未察觉到这种分布漂移。不确定性在筛选分子和testID分子之间的差异很小(KS统计量D=0.181,效果微弱)。如果只看不确定性,你会以为这140万个分子都在模型的操作范围之内——这显然是一个危险的误判。
而不熟悉度则清晰地揭示了强烈的分布漂移(KS统计量D=0.999,几乎是满分)。
图3 | 在140万个商业可得分子上的虚拟筛选。a,testID、testOOD与筛选库分子相对训练集的Tanimoto相似度分布。b,三组分子的预测不确定性分布(筛选库与testID差异很小,D=0.181)。c,三组分子的不熟悉度分布(筛选库与testID差异极大,D=0.999)。d,所有靶点上不确定性与不熟悉度的关系;e,针对PIM1激酶的对应关系。
这个对比极具说服力。同一批数据,不确定性视而不见,不熟悉度一目了然。这印证了之前文献的担忧:不确定性估计在强OOD数据上是不可靠的。
而且,不熟悉度揭示的分子洞察具有化学意义。高不熟悉度的分子往往结构非典型(例如一些奇怪的多卤代结构);低不熟悉度的分子则展现出生物活性分子的典型特征——类固醇骨架、已知的活性核心(例如针对PIM1激酶时,低不熟悉度的分子含有嘧啶酮等知名药效团)。
还有一个重要的排除:不熟悉度与药物相似性(QED)、合成可及性均无关联。这说明低不熟悉度并不是简单地在捕捉这个分子像不像药——它捕捉的是更深层的、与模型学到的分布相关的东西。而所有这些结构洞察,都是不确定性估计无法提供的。
搬进湿实验室的前瞻性验证
到这里,所有结论都还停留在计算层面。这篇论文最有分量的部分,是将方法搬进了真实的湿实验室。
作者针对两个有药理学意义的激酶靶点进行了前瞻性虚拟筛选:PIM1和细胞周期蛋白依赖性激酶1(CDK1)。特别值得一提的是,CDK1的数据完全未在研究中的其他任何地方使用过,是一个彻底独立的测试案例。
对每个靶点,作者使用全部可用数据训练了一个JMM(PIM1有1443个训练分子,CDK1只有312个),然后在约18万个药物样分子的商业库上预测活性。
分子的排序采用了一种多目标优化的方法——计算每个分子到乌托邦点(utopia point,多个目标的几何最优)的距离。三个目标分别是预测活性、预测不确定性、不熟悉度。作者故意设计了三种不同的权衡组合,以观察不确定性和不熟悉度在分布漂移下各自的表现:
方法A:高活性、高不确定性、低不熟悉度
方法B:高活性、低不确定性、低不熟悉度
方法C:高活性、低不确定性、高不熟悉度
为了将模型逼到训练分布之外,作者还额外设置了一道过滤:凡是与训练集或与已选分子的Tanimoto相似度大于等于0.70的化合物,统统排除。最终选出的60个分子(每个靶点30个,每种方法10个),全都与各自的训练集结构相距甚远——最大Tanimoto相似度仅约0.28。
实验结果(在10µM单浓度下测试):PIM1获得4个初步命中(抑制率大于50%)加6个弱命中(抑制率大于25%);CDK1获得1个初步命中加5个弱命中。
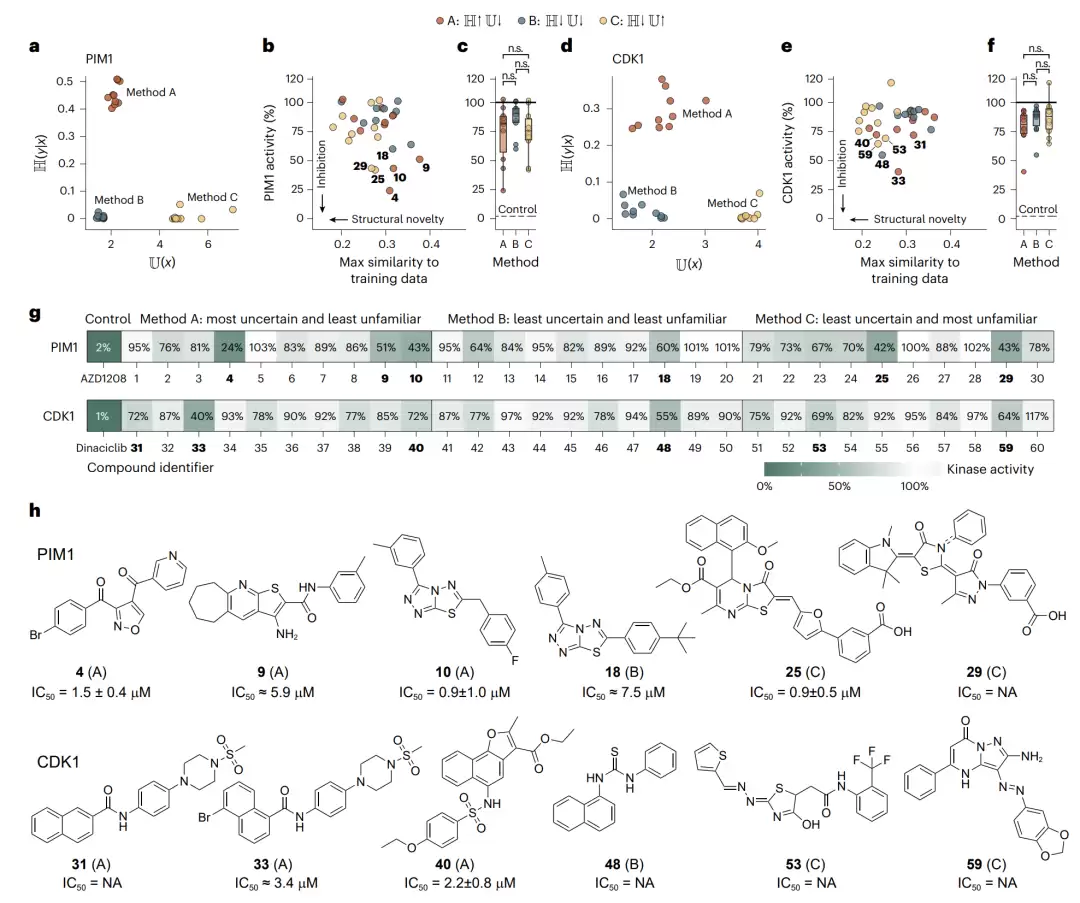 图4 | PIM1与CDK1的实验筛选。a与d,用三种不确定性-不熟悉度组合从约18万化合物中选出的十个最优分子。b与e,所选分子在10µM下的实测激酶活性及其与训练分子的最大相似度。c与f,各方法实测活性的箱线图。g,全部60个化合物的实测蛋白活性热图。h,各六个最有潜力化合物的结构与测定的IC50。
图4 | PIM1与CDK1的实验筛选。a与d,用三种不确定性-不熟悉度组合从约18万化合物中选出的十个最优分子。b与e,所选分子在10µM下的实测激酶活性及其与训练分子的最大相似度。c与f,各方法实测活性的箱线图。g,全部60个化合物的实测蛋白活性热图。h,各六个最有潜力化合物的结构与测定的IC50。
随后对每个靶点活性最高的6个化合物进行了剂量-反应曲线和IC50测定。PIM1的6个化合物均表现出剂量依赖的抑制,其中化合物4、10、25呈现清晰的S形部分抑制曲线,达到了低或亚微摩尔级的效力(IC50分别约为1.5µM、0.92µM、0.87µM)。CDK1这边由于训练集更小,抑制普遍较弱,仅有化合物40给出了完整的剂量-反应曲线(IC50约2.9µM)。
将这些数字放入背景中才能看出分量:本次筛选PIM1的命中率约17%,CDK1约7%。而传统的激酶聚焦筛选活动,命中率通常只有0.1%到5%。更难得的是,所有命中化合物与训练分子的最大子结构相似度均低于38%,而且完全来自纯前瞻性的、机器学习引导的选择。
最关键的一个观察:7个具有低微摩尔效力的化合物中,有5个来自选择方法A(低不熟悉度、高不确定性)。而依赖低预测不确定性来选分子的方法B和C,并未带来明显优势。这个结果意味深长,它说明在分布漂移下,低不熟悉度比低不确定性更能预示成功。
贡献与意义
退一步看,这篇论文真正提供的,是一个全新的视角来回答那个老问题:我们何时该相信一个分子机器学习模型的预测?
过去的两个答案——画相似度的圈(适用域)和看模型的置信度(不确定性)——各有各的盲区。不熟悉度补上了一块拼图:它通过模型重建分子的能力,同时捕捉了分子的结构距离和预测的可靠性。
更难得的是,它与不确定性是互补而非冗余的。两个指标各自都与性能相关,但彼此独立。这意味着你可以同时使用两者,获得对预测可靠性更立体的判断。
作者据此提出了几个明确的建议:用不熟悉度取代传统的、基于相似度的适用域定义;在筛选大规模分子库时采用不熟悉度,因为它能揭示那些被相似度忽略、被不确定性低估的分布漂移。更长远地看,由于不熟悉度的地形图实际上揭示了模型学到的分布中的空白区域,它有潜力指导强化学习,或者从一次性虚拟筛选拓展到主动学习这样的迭代式分子获取策略。
当然,这项工作也有其边界。湿实验验证的化合物数量不多,CDK1因训练集太小效果有限;命中的化合物虽然新颖且具有活性,但目前仍停留在微摩尔级,离真正的药物还有很长的路。但作为一个原理验证,它已经相当有说服力。
结语
回到开头那个悖论:我们最想要的,恰恰是模型最不擅长的。
这篇论文并没有消除这个矛盾,但它给了我们一个诚实的工具——让模型自己告诉我们,它对眼前这个分子到底有几分把握。在化学空间那片广袤的、尚未绘入地图的疆域里,这样一盏诚实的灯,也许比一个盲目自信的向导要珍贵得多。




