最近,化学合成领域传来一个令人振奋的消息。来自美国耶鲁大学的李昊特博士及其合作者,开发了一套名为MOSAIC的AI系统。这套系统的核心思路颇为巧妙:它将浩如烟海的化学合成知识,精细地划分成了2498个专业领域,并为每个领域专门训练了一个“专家”模型。
效果如何?在测试中,面对超过35种全新的化合物,MOSAIC给出的合成方案首次尝试成功率达到了71%。更让人惊讶的是,它甚至成功指导完成了此前文献中报道失败的化学反应,以及一些前所未见的新反应。你只需输入一个目标化学反应式,它输出的不是模糊的理论,而是一份可以直接送进实验室的操作指南——用什么试剂、加多少量、反应多久、如何提纯,乃至每一步的注意事项都清晰列明。这项重磅研究成果已正式发表于《自然》杂志。

图 | 李昊特
从化学家的困境出发
这项研究的起点,源于一个化学领域长期存在的根本性挑战。化学是一门高度依赖实验和经验的学科,其知识大厦建立在无数前人的试错与积累之上。这原本是学科的优势,但如今却带来了一个难题:每年发表的化学论文超过百万篇,没有任何一位化学家能够通读并记住所有文献中的细节。
从制药、新材料研发到催化与农业应用,新分子的合成往往意味着漫长的试错过程。一个有机反应的优化,通常涉及溶剂、浓度、加料顺序、温度与时间等多个相互影响的变量。探索一个新反应,耗费数月乃至数年时间是常态,其中的时间与资源成本可想而知。
正是为了应对这一困境,研究团队萌生了一个想法:能否构建一个系统,输入目标分子后,它不仅能判断能否合成,更能直接生成一套详尽、可靠、可立即执行的实验方案?
然而,现有的通用大模型虽然语言能力强大,但在处理具体化学反应时却常常“信口开河”。它们会用看似专业的术语编造一套方案,但试剂、温度、步骤往往错误百出。若真按其指导操作,很可能一无所获,甚至引发安全隐患。
(来源:《自然》)
“专家委员会”如何工作
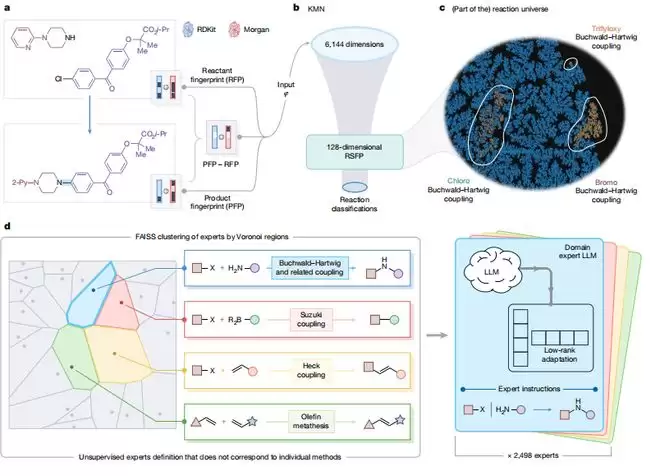
MOSAIC的解决思路独辟蹊径,它不像一个“通才”,而更像一个组织有序的“专家委员会”。具体来说,系统将整个化学合成知识空间分割成2498个区域,每个区域由一位“专家”负责。例如,某位专家可能专精于Buchwald-Hartwig偶联反应中特定氯代芳香底物的反应。
当输入一个新反应时,系统首先会在知识空间里定位,找到与它最“相似”的几位专家,然后请这些专家来给出方案。
那么,如何判断反应的相似性呢?研究团队首先训练了一个名为KMN的神经网络,它能将任何一个化学反应转化为一个128维的数字向量(称为“反应特异性指纹”)。两个反应的向量距离越近,就意味着它们在化学本质上越相似。
在此基础上,团队利用高效的检索工具FAISS,将整个知识库聚类成2498个“沃罗诺伊单元”,每个单元就是一位专家的“管辖范围”。在训练阶段,先让一个基础模型学习所有数据,再针对每个专家所在的细分数据集进行二次微调,使其成为该领域的“行家”。预测时,系统计算新反应的向量,找到最近的几个单元,激活对应的专家模型,最终整合输出完整的实验方案。
化繁为简的设计哲学
这条路并非一开始就如此清晰。团队最初也尝试过训练一个庞大的统一模型,但很快发现所需的计算资源(数百甚至上千张GPU)是大多数学术实验室无法承受的。
于是他们转换思路,从基础做起,只针对单一反应类型进行模型微调。结果发现,这种“化整为零”的策略反而取得了更好的效果。传统大模型方案需要庞大的算力支撑,而MOSAIC仅需几张GPU卡即可运行,并且具备可持续生长的能力。当有新数据加入时,无需重新训练整个系统,只需在知识空间中新增“沃罗诺伊单元”,训练新的专家即可,原有专家不受影响。检索时,系统会同时查询新旧索引,合并排序后给出结果。这种去中心化的设计,对资源有限的学术界极为友好。
不只是模仿,更能创新
性能是检验系统的唯一标准。研究人员用MOSAIC预测了37种新化合物的合成路线,其中35种在第一次实验尝试中便获得了成功。
一个尤为突出的案例是某种5-氮杂吲哚衍生物的合成。这类化合物在既往文献中被标记为“难以用现有方法制备”。MOSAIC给出的预测方案,其与知识库中最近专家中心的距离高达320,远超过通常150的置信阈值。这意味着,该反应在已有知识库中几乎找不到先例。
然而,研究人员完全依照AI的预测进行实验,竟然成功获得了目标产物。分析表明,MOSAIC找到了一种此前未被报道过的环化方法。这证明该系统并非简单地照搬和组合已有知识,而是在知识的边缘地带具备了一定的泛化与创新能力。论文审稿人也对此特别赞赏,认为实现全新反应的预测是本研究的一大亮点。
可量化的“置信度”:从黑箱到导航仪
另一个关键贡献在于,MOSAIC为它的预测提供了一个可量化的“置信度”指标。研究人员将所有合成尝试的结果与预测距离进行对比,发现了一个清晰的规律:当预测距离小于100时,实验成功率超过75%;当距离大于200时,成功率则降至50%左右。
这个置信度指标成为了实验优先级排序的有效工具。高置信度的预测可以大胆尝试,低置信度的则可以作为探索方向,但需要预留更多容错空间。审稿人认为,这弥补了一个长期存在的瓶颈——过去只有领域专家才能评估AI预测的可靠性,而现在有了一个客观的量化尺度。
此外,研究还发现“群策群力”的优势。在预测试剂和溶剂时,单个专家模型的精确匹配率分别为22.4%和29.8%。但如果让三位专家共同“投票”决策,精确匹配率几乎翻倍,分别提升至43%和32.8%,部分匹配的成功率更是达到了94.8%。这说明集成多位专家的意见,能显著提升预测的可靠性。

(来源:《自然》)
背后的思考与未来展望
李昊特博士在分享中提到,研究初期团队内部也曾有过疑虑。当时已有类似Chemcrow这样的架构,通过给GPT-4等大模型发送指令来完成化学任务,似乎已经涵盖了他们的目标。但团队从不同角度深入审视后,发现现有商业模型在深层化学理解上仍有巨大提升空间,并通过实验数据证实了这一点。这个插曲让他们深刻体会到,在科研中保持独立思考、不满足于“可行方案”而追求“更优解”的重要性。
当然,MOSAIC目前并非完美。它最大的局限在于“黑箱”特性——虽然能以高概率给出成功方案,但尚不能解释自己为何做出这样的预测。“可解释性”已成为团队下一步的研究重点。未来的目标不仅是给出方案,还要能说清楚选择该反应条件的内在逻辑,这才是让AI真正“理解”化学的关键一步。
展望未来,这项技术在药物合成、新材料发现等领域具有广阔的转化前景。最直接的应用便是与全自动机器人合成平台结合,形成“AI设计-机器人执行”的闭环,极大加速新药与新材料的探索进程。
此外,MOSAIC也可以与现有的Reaxys、SciFinder等大型化学文献数据库结合。过去在这些平台上搜索一个反应,可能返回海量相似文献,让人无从筛选。MOSAIC能够将这些信息整合、提炼,直接生成最精简、最可能成功的合成路线。在实验室里,时间是最昂贵的成本,MOSAIC的价值就在于将化学家从翻阅成千上万篇文献的繁重劳动中解放出来,将方向筛选的时间缩短到几分钟。
目前,研究团队已经将MOSAIC项目开源,相关代码和模型均已公开,供学术界和工业界的研究者使用、测试与发展。
参考资料:
相关论文 https://doi.org/10.1038/s41586-026-10131-4




