蛋白质结构基序在本质上可定义为蛋白三维空间中那些反复出现的局部构型单元,通常由若干关键残基组合而成。这些看似微小的残基却往往扮演着稳定性锚点、配体结合口袋乃至催化活性核心的重要角色。过去的功能注释高度依赖序列比对,但序列与功能之间并非简单的线性关系。尤其在远缘同源蛋白中,序列相似性可能已完全消失,而功能相关的结构基序却依然保持高度保守。
随着AlphaFold2及各类蛋白质语言模型的突破性发展,研究人员掌握了数以亿计的预测结构,堪称结构生物学领域的“数据盛宴”。然而,随之而来的挑战同样显著——如何在浩瀚的结构宇宙中快速定位相似的结构基序?这无疑是一个极具难度的课题。
近期,一款名为Folddisco的结构基序搜索工具提供了一个令人印象深刻的新方案。其核心思路巧妙:采用位置无关的几何特征索引,结合侧链方向信息与基于稀有度的评分机制。实际表现相当令人振奋——查询速度提升约20倍,存储需求降低4倍,且准确率更优。最值得关注的是,Folddisco能在5300万个蛋白质结构中实现秒级搜索,不仅支持短基序,就连长距离、非连续的结构片段也能轻松应对。这为功能注释、催化位点发现、蛋白质互作研究乃至构象状态识别打开了全新局面。

事实上,蛋白质结构基序这一概念由来已久。它们是三维空间中反复出现的“标准模块”,通常由少量关键残基构成,承担着金属离子结合、催化反应、受体激活等核心功能。有趣的是,即便两条蛋白质的序列已发生极大分化,只要保留关键的结构基序,它们仍可能执行类似的生物学任务。
长期以来,蛋白质功能注释主要依赖序列比对方法,但这一途径存在根本局限——序列与功能之间并非直接对应关系。尤其是远缘同源蛋白,序列可能已演化得毫无相似之处,而功能相关的结构基序却依然保持稳定。
近年来,AlphaFold2等工具使蛋白质结构预测变得高效且可规模化,数以亿计的高质量模型迅速涌入公共数据库。结构搜索由此成为功能预测的新兴路径。不过,像Foldseek这样的工具虽能实现大规模结构比对,但其主要关注整体结构相似性,面对结构基序中常见的非连续残基匹配问题常显得无能为力。
至于早期的RCSB Motif Search和pyScoMotif,它们确实具备局部结构基序搜索能力,但缺点也十分明显——索引构建速度缓慢、存储开销庞大,且处理长距离或复杂结构基序时效果不佳。因此,开发一款兼具高精度、高效率与高扩展性的结构基序搜索工具,已成为一个切实的迫切需求。
方法
Folddisco的处理方式是,首先分析蛋白质结构中彼此接近的残基对,并为每一对提取七类几何特征。这七类特征包括:氨基酸类型、Cα原子距离、Cβ原子距离、侧链夹角,以及两种能反映侧链朝向的二面角。随后,这些特征被编码为32位整数,构建成倒排索引数据库。
查询阶段,系统根据输入的基序提取相应特征,通过索引快速筛选出候选结构。接着,基于逆文档频率(IDF)的覆盖度评分被用于评估匹配质量——对稀有特征给予更高权重。通过预筛选的候选蛋白质进一步构建残基匹配图,识别出能组成完整结构基序的残基集合,并通过三维叠合计算结构偏差,最终确定匹配结果。
此外,研究人员还引入了允许氨基酸替换、距离容差及角度容差的扩展搜索策略,从而在面对结构变异和远缘同源蛋白时,依然能保持强大的检索能力。
结果
Folddisco 实现蛋白质宇宙尺度的结构基序搜索
研究人员首先搭建了Folddisco的整体框架,并在由5300万个蛋白质结构组成的AFDB50数据库上进行了测试。
与RCSB Motif Search和pyScoMotif相比,Folddisco采用了“无位置索引”设计——仅记录结构编号,不保存残基位置。这一策略显著降低了存储需求。同时,新增的两个二面角特征有效描述了侧链方向信息,提升了结构识别能力。
最终,Folddisco仅用不到25小时便完成了5300万个蛋白质结构的索引构建,数据库大小约为1.45 TB。而现有方法则需要4倍以上的存储空间,耗时也更长。
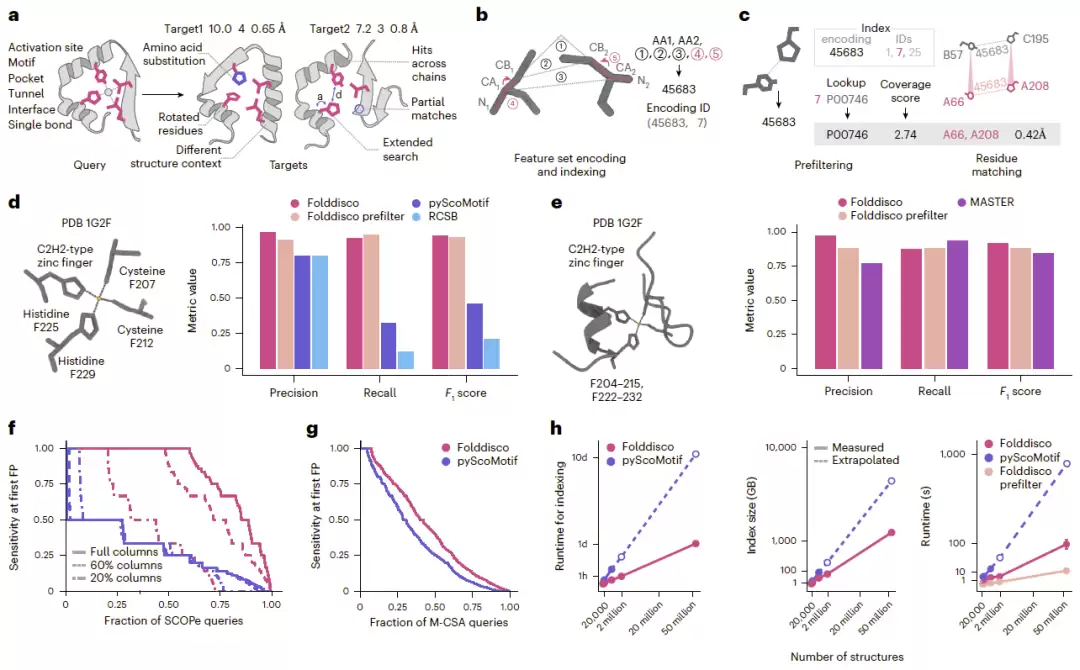
图1:Folddisco工作流程与性能评估示意图。
结构基序识别准确率显著优于现有方法
研究人员首先使用人类蛋白质组中的锌指结构与丝氨酸蛋白酶催化三联体作为测试案例。
对于简单的三残基锌指基序,Folddisco、RCSB和pyScoMotif的表现不相上下。但换成完整的四残基C2H2锌指结构后,Folddisco的优势便显现出来,召回率和F1分数均显著更高。
再与MASTER相比,Folddisco不仅能识别短基序,对较长且不连续的结构片段同样有效。搜索包含锌指区域的结构片段时,Folddisco准确率更高,速度提升7倍;若仅使用预筛选模块,速度提升甚至超过1700倍。可以说,Folddisco是目前唯一能够同时处理离散结构基序和长距离不连续结构片段搜索的方法。
在SCOPe和M-CSA基准测试中展现更强泛化能力
为验证该方法的普适性,研究人员构建了一套基于SCOPe数据库的结构家族基准测试。他们从多个结构家族中提取保守残基,构成模拟基序,然后要求模型在整个SCOPe数据库中检索属于同一家族的蛋白质。
结果显示,无论使用完整保守位点还是仅保留部分位点,Folddisco的表现都明显优于pyScoMotif。使用完整保守位点查询时,Folddisco的AUC达到0.837,而pyScoMotif仅为0.285。
随后,利用M-CSA数据库中人工整理的催化位点进行测试。Folddisco的AUC为0.432,比pyScoMotif提高25.6%。切换至高灵敏度模式后,AUC进一步提升至0.463。这些结果充分说明,Folddisco不仅能识别简单的结构基序,对于复杂的催化位点及远缘同源功能位点同样表现优异。
速度提升20倍,存储开销降低4倍
研究人员对Folddisco的扩展能力进行了系统评估。在包含54万个蛋白质结构的数据库上,Folddisco仅用18分钟便完成索引构建,而pyScoMotif需要3.46小时。
索引存储方面,Folddisco仅占用23.2 GB,pyScoMotif则需要79 GB。将这个比例外推至5300万个结构的AFDB50数据库,Folddisco大约需要1.45 TB存储空间,而pyScoMotif预计需要5.7 TB。
查询速度方面,Folddisco的完整搜索流程比pyScoMotif快18至20倍;仅使用预筛选阶段时,速度提升可达86至130倍。即便面对AFDB50这一规模,单次查询也只需大约12秒即可完成。
这些数据清楚表明,Folddisco首次实现了真正意义上的“蛋白质宇宙级”结构基序搜索。
成功发现远缘蛋白中的保守功能基序
研究人员还在真实生物学问题中展示了Folddisco的能力。以经典的C2H2锌指结构作为查询对象,Folddisco成功在太平洋牡蛎的未注释蛋白以及环境宏基因组蛋白中发现了完整的锌指结构——而此前,这些蛋白均缺少InterPro等数据库的注释。
更有趣的是,在大肠杆菌的肽脱甲酰酶中,Folddisco识别出部分锌指结构对应的金属结合位点,与实验已知的功能位点高度一致。相比之下,Foldseek对这些局部结构基序束手无策,匹配显著性很低,甚至完全无法比对。
这一结果表明,即使蛋白质序列已分化得面目全非,Folddisco仍能通过结构基序发掘出隐藏在深处的功能关联。
识别蛋白构象状态与蛋白互作界面
研究人员进一步测试了Folddisco对蛋白质功能状态的识别能力。使用GPCR受体激活相关的三个经典基序——CWxP、NPxxY和DRY——作为查询对象,结果Folddisco能准确区分活化态与非活化态的受体结构。
在PDB数据库中,54%的匹配结果属于活化态;在AlphaFold数据库中,这一比例为53%。这表明,AlphaFold预测结构中真实生物体系中的构象分布特征得到了良好保留。
此外,研究人员以免疫球蛋白结构域的界面作为查询对象,在AFDB50数据库中成功检索到具有相同结合几何特征的单链抗体片段。
更进一步的测试显示,对于同时包含活性位点与变构位点的蛋白质,Folddisco能在82.9%的情况下同时识别两类位点,证明其具备处理复杂功能基序的能力。
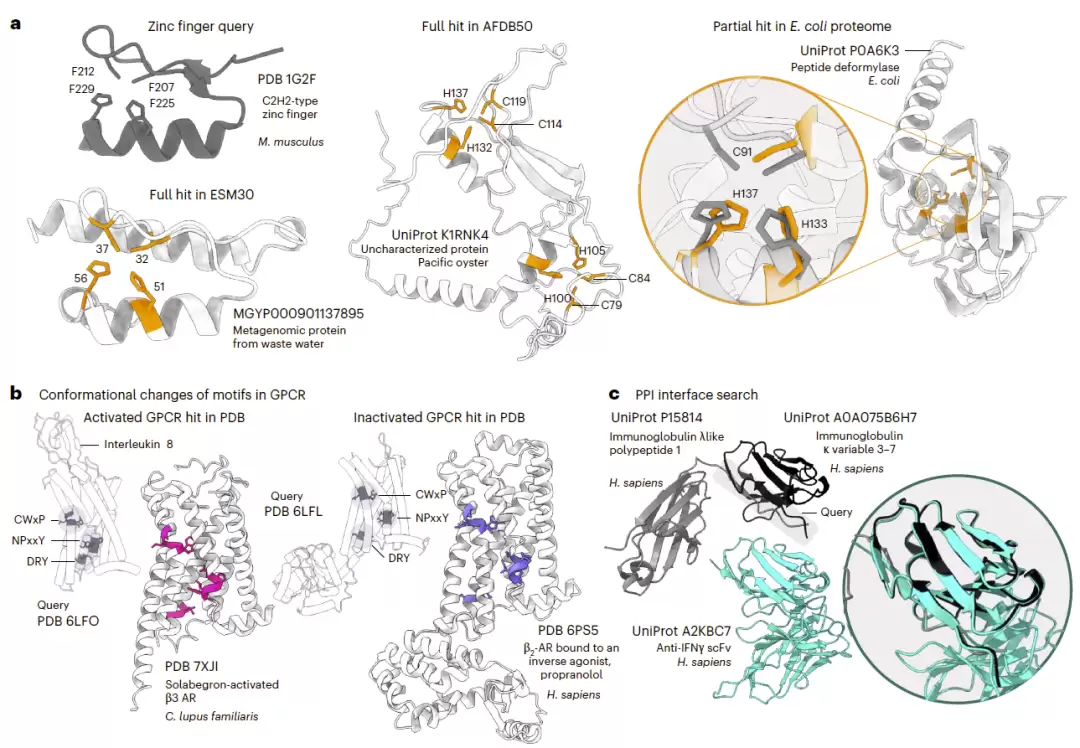
图2: Folddisco在功能位点、构象状态及蛋白互作界面中的应用示例。
讨论
研究人员开发的结构基序搜索工具Folddisco,使蛋白质宇宙尺度的高效结构检索成为现实。通过引入侧链方向几何特征、无位置索引结构以及基于稀有度的覆盖评分机制,Folddisco在准确率、查询速度和存储效率上均明显优于现有方法。
与传统结构比对工具不同,Folddisco不仅能识别短催化基序和金属结合位点,对长距离、不连续的结构片段以及蛋白质互作界面也能轻松处理。这使其成为连接蛋白质结构与功能的重要桥梁。
当然,研究人员也指出,当前方法仍存在一定局限。例如,固定距离分箱可能遗漏边界匹配,连接图约束使超过20 Å的远距离功能位点难以识别,而短结构基序的排序策略也有优化空间。
未来方向是什么呢?研究人员计划引入专门的E-value统计模型和动态分箱机制,并进一步扩展到核酸结构、蛋白质-配体相互作用,甚至AlphaFold 3预测的生物大分子复合体。最终目标是构建一个覆盖整个生物分子宇宙的结构基序搜索平台。
参考资料
Kim, H., Kim, R.S., Mirdita, M. et al. Structural motif search across the protein universe with Folddisco. Nat Biotechnol (2026).
https://doi.org/10.1038/s41587-026-03162-9





