Claude AI化学家:结构谱图双向推导
时间:2026-06-08 16:17
Anthropic测试Claude模型在核磁共振谱图预测与结构解析中的能力。Opus4 7在正向预测(结构→谱图)中与专业软件相当或更优,平均氢谱误差±0 079ppm;逆向推断(谱图→结构)中,模型仅凭分子式和谱图即可正确还原简单分子结构,实现了传统软件无法完成的自动解析。
一、看不见的分子,与化学家的多重语言
化学领域存在一个外人难以理解的事实:分子实在太小了,即便最顶尖的显微镜也无法直接观察到它的具体形态。当一位化学家完成一步反应后,他无法凭肉眼判断烧瓶里到底生成了什么。只能借助各种能量——可见光、射电波、磁场——去探测这个分子,再根据分子对这些能量的吸收、发射或偏转方式,反推出它的结构。
更棘手的是,同一个分子在化学家的日常工作中会以完全不同的面貌出现。白板上的手绘结构式、仪器打印出来的谱图、数据库查询时输入的字符串、专利和论文中严谨的系统命名……每一种表示形式背后都是同一套化学,但每一种都需要不同的解读方法。
文章举了一个很妙的例子:当化学家看到咖啡因的结构草图,他能一眼认出它与腺苷(adenosine,即身体里负责让人犯困的信号分子)高度相似,从而推测咖啡因之所以提神,是因为它抢先占据了腺苷本该结合的受体。但同样这张草图却无法帮助他把咖啡因与其他几个看起来几乎一模一样的分子区分开来。不同的任务,需要不同的表示方式。
为什么准确识别分子如此重要?因为化学几乎是所有事物的底层——我们吃的食物和药物、涂抹的护肤品、使用的油漆和塑料,都建立在分子之上。而分子对结构的微小差异极其敏感。同样数量的原子,只需重新连接几根化学键,葡萄糖就会变成果糖:分子式完全一致,但在体内的代谢通路却截然不同。更极端的例子是,将某个分子翻转为其镜像,一种镇静剂就可能变成致畸剂——这正是当年沙利度胺(反应停)悲剧的根源,一种用于治疗孕吐的药物,最终与全球超过一万名儿童的严重出生缺陷联系在一起。
而这件事的规模大到令人绝望。全球最大的化学物质登记机构CAS,已经收录了超过2.9亿种公开披露的物质,并且每天仍在以约1.5万种的速度增长。在不同表示方式之间来回翻译——从一张图中提取结构、将仪器读数与设想产物对应、用正确的符号去查询数据库——既耗时,又根本不可能靠人力跟上这样的增长速度。
二、为什么是现在
先澄清一个说法:
AI能帮助化学家这件事,并不是一个新想法。
机器学习被寄予厚望已有多年,尤其是在逆合成分析(即从一个目标分子倒推,规划出该用哪些更简单的原料、按照什么步骤合成出来)、反应预测和性质估算等方向。但这些工具所需的数据一直难以获取:阴性结果(即失败的实验)几乎无人记录、数据格式五花八门、大量内容被锁在付费期刊和非结构化的补充材料中。逆合成就是一个典型案例——可用的AI工具其实早已存在,但普及程度极不均衡,普通高校实验室或小型课题组的化学家往往根本用不上它。
那为什么是现在?因为前沿模型已经发生了质的飞跃。
当今的模型是多模态的,并且能够进行显式推理。它可以直接从一篇论文的插图、甚至一张手绘草图中读出化学结构,而无需依赖事先整理好的分子数据库;它可以读懂方法部分和补充材料中那种真实发表出来的、凌乱的实验细节;最关键的是,它能把推理过程一步步摊开给你看,这意味着化学家可以审查它的输出,而不是面对一个黑箱。
这些并没有消除化学界念叨多年的数据难题。但它改变了一点:在数据不足的前提下,哪些问题变得可解了。Anthropic对自己的定位相当克制——Claude正开始在那些与化学家判断力互补的日常工作上,也就是翻译、回忆与整合方面,提供有意义的帮助,而他们打算将这种帮助继续向前推进。
三、第一道考题:核磁共振谱
作为整个计划的第一篇白皮书,他们选择了核磁共振(NMR)谱图作为切入点。这个选择非常合理:NMR是化学家进行结构确认时最常用、也最耗时的技术之一。
简单解释一下这个过程。化学反应几乎从未干干净净只生成一种产物,而确认所得到的化合物正是你想要的那个,往往要消耗化学家大半的表征时间。NMR的做法是把样品放入强磁场,再用射电波去激发它:分子中每个化学环境不同的氢原子或碳原子,都会在略有不同的位置上给出一个信号峰。最终你得到的是一排峰——本质上是这个分子的指纹。而所谓读谱,就是把谱图上的每一个峰人工对应到结构中的某一个原子。这是合成化学中最磨人的步骤之一。
实验的设计本身就值得关注。研究者用三个Claude模型(Opus 4.7、Opus 4.6、Sonnet 4.6),对阵化学家桌面上几乎人手一份的两款专业软件:ChemDraw和MestReNova。测试使用了20个化合物,而这20个化合物有一个关键来源——它们全部取自ChemRxiv上、在这些模型训练数据截止之后才发表的预印本。
这一点很重要,值得专门强调。把测试材料限定在模型训练截止日期之后,等于堵死了一种最常见的质疑:模型不是真的会计算,只是早就把答案背下来了。这些分子是全新的,模型在训练时不可能见过它们的谱图,所以它的表现只能来自推理,而不是记忆。同时,研究者还特意在生成任何预测之前,就把化合物选定并锁死,以避免选择偏差。这是非常扎实的实验设计。
这20个化合物分为四个结构家族,每个家族5个,每一族都被刻意挑选来代表一类不同的NMR难题。第一族是含有缓慢交换NH质子的氯哒嗪,这类质子的位置出了名的难以预测;第二族是马来酰亚胺与炔酰胺,考验的是一类特殊的羰基,以及炔酰胺中那对罕见的α、β碳;第三族是带有非对映异位CH₂的螺环酮;第四族是α-硅基甲磺酰胺,其中紧邻硅原子的那个碳会被强烈屏蔽。换句话说,这并非一份挑软柿子捏的考卷。
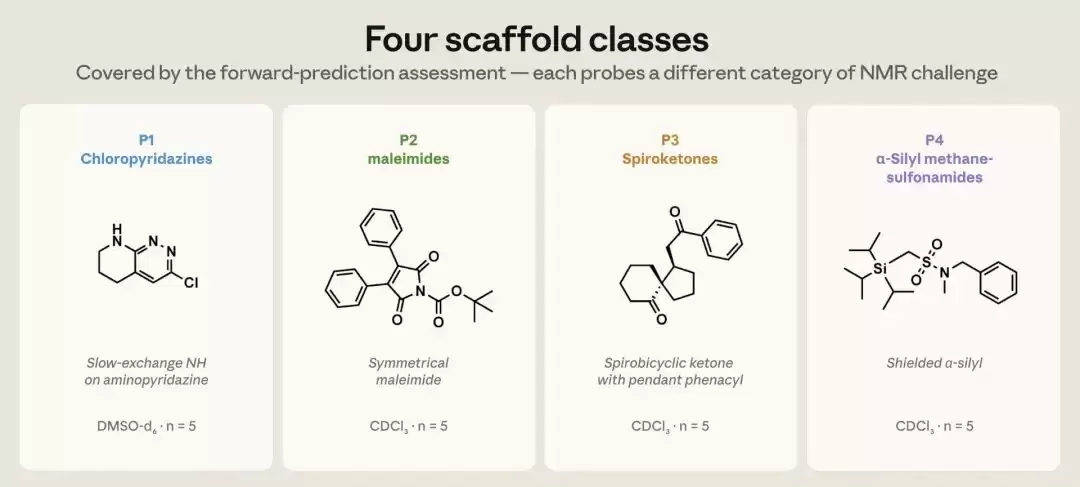
图1 正向预测评测覆盖的四类结构骨架,每一类对应一种不同的 NMR 难题——氯哒嗪(P1)、马来酰亚胺与炔酰胺(P2)、螺环酮(P3)、α-硅基甲磺酰胺(P4),每类 5 个、共 20 个化合物。(图片来源:Anthropic)
图1 正向预测评测覆盖的四类结构骨架,每一类对应一种不同的 NMR 难题——氯哒嗪(P1)、马来酰亚胺与炔酰胺(P2)、螺环酮(P3)、α-硅基甲磺酰胺(P4),每类 5 个、共 20 个化合物。(图片来源:Anthropic)
测试分为方向相反的两道大题:正向预测(从结构推谱图)和逆向预测(从谱图推结构)。后者要难得多,也恰恰是现有软件留给化学家自己去啃的那一块。
四、正向预测:和专业软件正面硬碰
正向预测是这两件事中比较常规的那一个:你画出预想的结构,让工具预测它应该呈现出什么样的谱图,再拿去与实测结果对比。ChemDraw和MestReNova做的就是这件事。
考虑到大模型每次输出都会有些波动,每个Claude模型对每个化合物都被询问了三遍并取平均值;而两款软件是确定性的,每次答案都一样,所以只运行一遍。然后研究者将每一个预测峰与其对应的实测峰配对,计算出两者相差多少ppm(化学位移的单位)。化学家眼中算正确的窗口是:氢谱 ±0.20 ppm,碳谱 ±1.0 ppm。
结果如下。在氢谱上,Opus 4.7是所有工具中最准确的,平均误差仅为±0.079 ppm——还不到容差窗口的一半——落在窗口内的峰的比例也是最高的。在碳谱上,Opus 4.7(±1.37 ppm)和MestReNova(±1.48 ppm)基本持平。Opus 4.6不出意料地处于中游,Sonnet 4.6最弱。
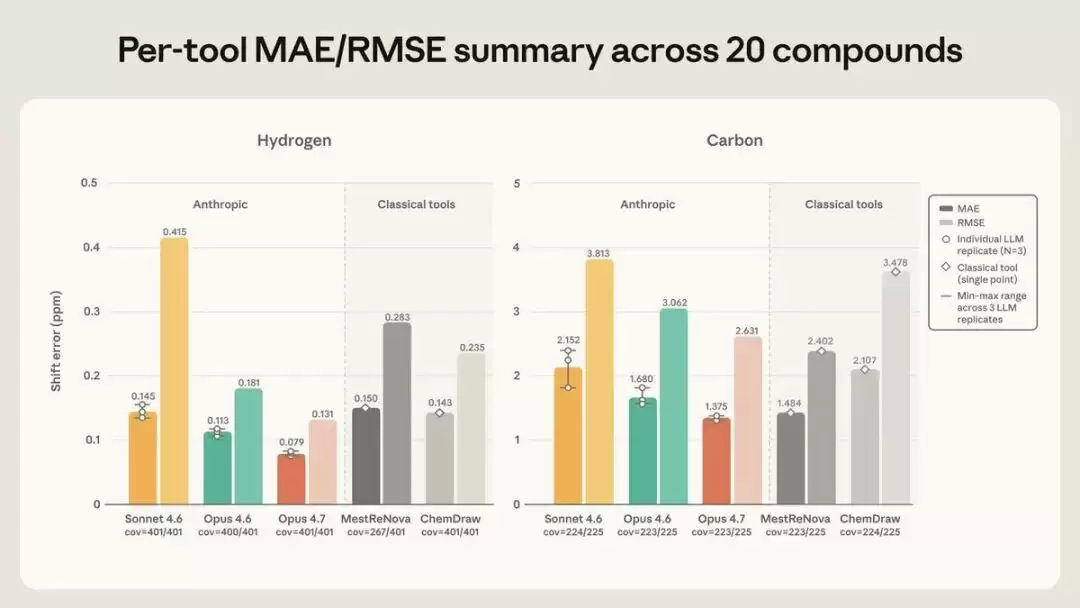
图2 20 个化合物上各工具的氢谱(左)与碳谱(右)位移误差,深色为平均绝对误差 MAE、浅色为均方根误差 RMSE,下方标注峰覆盖率。Claude 取三次重复的均值并标出最小到最大范围,传统软件为单次确定性预测。(图片来源:Anthropic)
图2 20 个化合物上各工具的氢谱(左)与碳谱(右)位移误差,深色为平均绝对误差 MAE、浅色为均方根误差 RMSE,下方标注峰覆盖率。Claude 取三次重复的均值并标出最小到最大范围,传统软件为单次确定性预测。(图片来源:Anthropic)
模型之间的差异,在一个出了名难缠的氢质子上体现得最为明显:氯哒嗪家族中的那个NH质子,其真实位置落在6.8到7.9 ppm这个很窄的区间内。Opus 4.7将其预测得略低,但稳定地偏低;Opus 4.6的几次猜测散落在好几个ppm上;而Sonnet 4.6干脆把它扔到了10到13 ppm,离实际位置差了十万八千里。这个细节很说明问题——同一个体系,能力的差异并不体现在平均分上,而在于它是否会在最困难的地方彻底跑偏。
更有趣的是峰形和峰距。一个氢的信号会分裂成什么形状(单峰、双峰、三峰……),以及裂开的小峰彼此间隔多远,这些里面同样包含着化学家需要读取的结构信息。在裂分模式上,Opus 4.7与实测结果匹配的次数比任何其他工具都多。而在描述小峰间距的耦合常数(J值)上,三个Claude模型大约有80%的预测能精确到半个赫兹以内——而ChemDraw和MestReNova的这个比例只有26%到35%。这里的差距并非细微领先,而是数量级上的碾压。Opus 4.7同时也是三次重复中最稳定的,它自己几次之间的平均误差波动,比它领先第二名的幅度还要小。
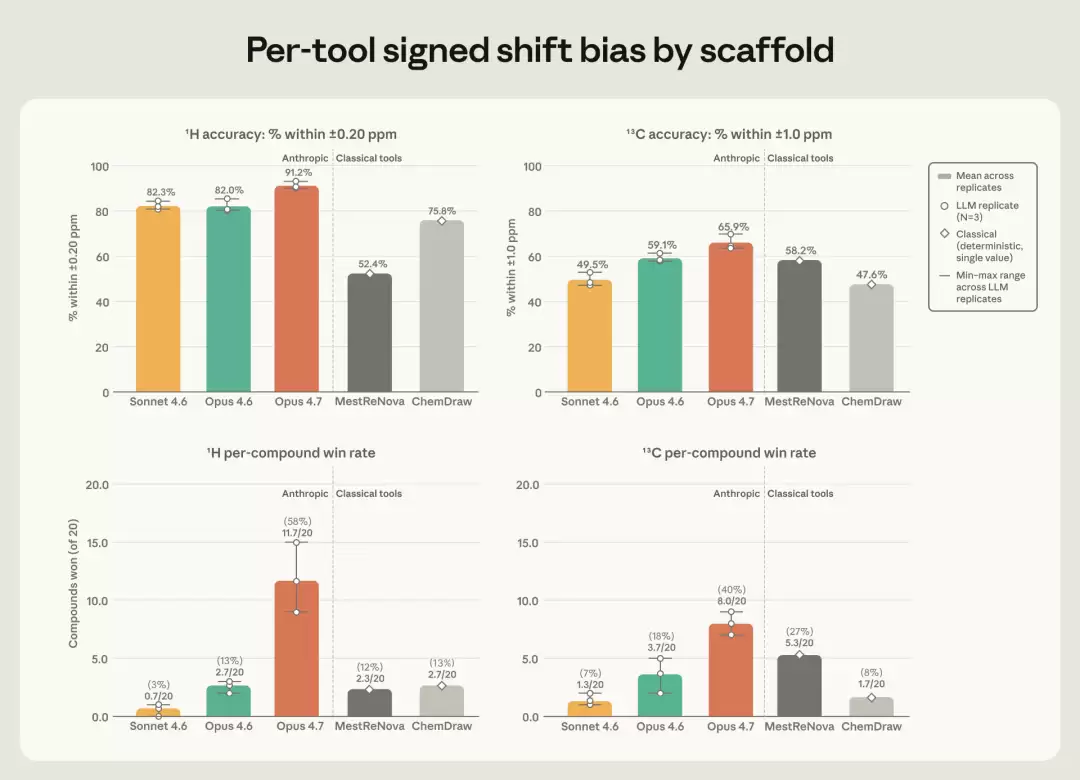
图3 上排为落在容差窗口内的原子比例(氢谱 ±0.20 ppm、碳谱 ±1.0 ppm);下排为逐化合物胜出率,即在 20 个化合物中,每个工具拿到最低单化合物 MAE 的次数。(图片来源:Anthropic)
图3 上排为落在容差窗口内的原子比例(氢谱 ±0.20 ppm、碳谱 ±1.0 ppm);下排为逐化合物胜出率,即在 20 个化合物中,每个工具拿到最低单化合物 MAE 的次数。(图片来源:Anthropic)
当然,专业软件也有它的主场。ChemDraw最大的长处是覆盖面——它在氢谱和碳谱两边都保持着最广的峰覆盖率,哪怕它的耦合常数计算不准。换句话说,它什么都预测,只是不一定都准;而Claude预测得更准,但偶尔会漏掉一些原子。这是一个很真实的权衡,而非一边倒。
五、逆向推断:去做软件做不到的事
如果说正向预测是Claude去与专业软件比拼谁更准确,那么逆向推断就是Claude去完成一整类软件根本做不到的事情——这才是这篇白皮书里真正令人眼前一亮的所在。
逆向推断,也称为结构解析:给你一张谱图,反过来确定背后究竟是什么结构。这需要专家级的推理——要判断分子中有哪些片段,以及它们是如何连接起来的。ChemDraw完全没有这个能力;MestReNova能帮你把峰归属到一个已知结构上,但它无法从一张峰列表凭空生成候选结构。这一步,传统上一直是留给化学家本人的。
研究者给Opus 4.7提供了15道结构解析题,每题做三遍,要求它给出最多三个排序后的候选结构。每道题提供的是该化合物的精确分子式(来自高分辨质谱HRMS)以及它的氢谱和碳谱。这15道题按难度分为两档:8道较简单的(单环或两片段的分子),仅给分子式和谱图;7道更复杂的(稠环、螺环等),额外多给一个提示——那个投入反应中的起始原料的结构。
值得说明的是,除了那一点点额外提示外,研究者刻意不给任何其他反应背景:没有试剂、没有条件、没有机理,也没有告知产物属于哪一类。这实际上模拟了化学家真实会遇到的两种情境——确认一个来源不明的反应的产物,以及确认一个已知投料的反应的产物。
结果相当亮眼。那8道较简单的题,Opus 4.7仅凭分子式和谱图,每一次尝试都正确还原了结构。在7道更难的题上,借助起始原料这个提示,它对其中4道在三次尝试中全部答对,剩下几道也在三次中答对了两次。
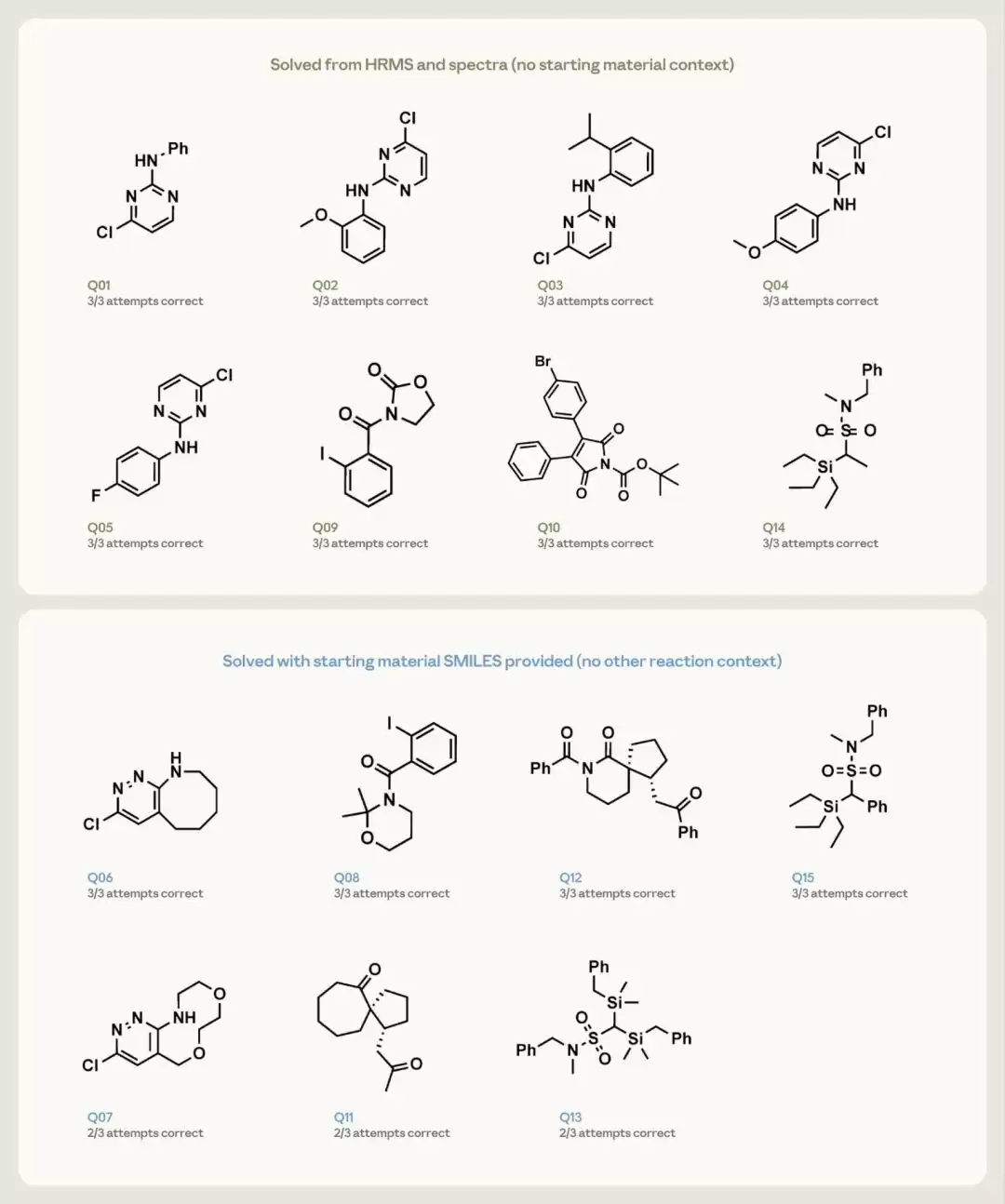
图4 15 道逆向结构解析题的结果,每格标注该题三次尝试中答对的次数。绿色边框表示只给谱图和高分辨质谱、不给起始原料;蓝色边框表示额外提供了起始原料的结构(但不含任何其他反应信息)。(图片来源:Anthropic)
图4 15 道逆向结构解析题的结果,每格标注该题三次尝试中答对的次数。绿色边框表示只给谱图和高分辨质谱、不给起始原料;蓝色边框表示额外提供了起始原料的结构(但不含任何其他反应信息)。(图片来源:Anthropic)
为什么这件事重要?因为专门的结构解析软件其实已经存在了几十年,但它通常需要二维核磁(一种有两个坐标轴、输出是等高线图而非一排峰的谱图)、需要专门的训练、还需要付费授权。而Claude所用的,就是化学家会直接粘贴进对话框里的那些内容——一张常规高分辨质谱和一份一维峰列表,无需任何额外的准备。它把自动结构解析从原本需要依靠二维实验或人工硬解的领域,向前推进到了仅凭手头现有的一维数据就能完成的程度。
六、一份诚实的边界清单
这篇白皮书值得关注的一点是,它对自己的局限交代得相当坦白,没有把一个初步结果包装成一场革命。
最直接的方面:这次评测规模很小——正向任务20个化合物、四个骨架,逆向任务15个。而且每一类骨架只贡献了一种失败模式,因此这些数字排名应被视为具有指示意义,而非精确结论。
第二,在最密集的那几道逆向题中,如果不提供起始原料,模型可能会在推理中反复绕圈、迟迟不肯锁定一个最终结构——这正是那7道难题为什么要附上起始原料而非只给谱图的原因。这是一个非常诚实的承认:在最困难的情况下,模型仍然需要一根拐杖。
第三,有些化学骨架根本没有被测试到。例如前面提到的缓慢交换的NH芳杂环,这次仅通过氯哒嗪取样,像羟基吡啶、氨基噻唑等相关体系均未覆盖。第四,二维实验(COSY、HSQC、HMBC)和立体化学从设计上就被排除在外,因为一维核磁本身就无法确定构型——也正因如此,复杂的天然产物没有参与评测。第五,溶剂只覆盖了三种,甲醇、苯、丙酮等常用氘代溶剂均未被评估。
还有一个相当有趣的共性瑕疵被特意指出:所有工具——包括两款软件和Claude——都会把羰基碳预测得偏低一些。这意味着如果你想通过两个工具是否一致来对羰基碳进行交叉验证,是无法发现这个错误的,因为它们会一起出错。这种对系统性偏差的洞察,恰恰是认真做评测才会注意到的东西。
研究者也提到了理想状况下应该怎么做:评测应扩展到几百个化合物、覆盖20到30类骨架、每类至少15个,这样才能将同一类内部的波动与工具之间的真实差异区分开来;还应该补充那些未测试的NH芳杂环、那些未测试的溶剂,并制作出动用二维实验的版本。
七、路线图,以及这件事真正的分量
NMR只是一个开端。文章列出了接下来要重点攻克的几个瓶颈,都是最拖慢化学家的环节。一是读取和渲染化学结构,即把图、专利、幻灯片或草图中的结构转换为机器可读的形式,并在结构表示与文献中的系统命名之间自由换算。二是反应与合成推理,包括提出、评估、批判一条合成路线,预判结果,理清选择性、条件和可能的副产物。三是机理,用化学家真正使用的语言——电子箭头、中间体、过渡态——去解释和检验反应机理。四是化学文献理解,去读懂文献中那种同一个分子可能被画出来、被命名、被缩写、或被一个代号指代的真实写法,并从方法部分、补充材料和专利中把真正重要的化学信息提取出来。
值得注意的是,这几件事并不在同一条成熟度曲线上。光谱分析已经成熟到可以拿来做基准测试了,而像逆合成规划这样的方向,还停留在被界定范围的阶段。Anthropic表示,随着对这些瓶颈的理解越来越清晰,他们会持续公布当前模型在哪些地方表现出色、又在哪些地方仍然不足——最终目标是让一线化学家清楚地知道,Claude在哪里能帮他们节省时间,又在哪里仍然需要依靠自己的专业判断。
读完整篇文章,有几个值得深思的要点。
第一,最反直觉的事实是没有专门微调。Opus 4.7是一个通用模型,它没有为化学领域单独进行过精调,却能在一项高度专业的任务上追平甚至局部反超用了几十年的专用软件。这与过去那种为每个垂直领域训练一个专门AI的范式截然不同——它暗示通用推理能力本身,正在跨越越来越多原本被认为需要专用工具的门槛。
第二,正向和逆向这两道题,分量其实不对等。在正向预测上,Claude是在与已有软件比拼谁更准确,这是一种追平;但在逆向解析上,Claude做的是这一类正向预测软件在原理上根本做不到的事,这是一种新增的能力。前者令人佩服,后者才真正改变了化学家能向一个模型提出什么样的请求——确认一个已知反应的产物、排除一个区域异构体、对峰归属进行一次快速核验、或者判断哪些化合物值得再去做二维实验,这些原本要么得靠专业软件、要么得靠专家脑子的工作,现在用纯文本对话就能处理了。
第三,也别忘了那个被忽视的普通用户。文章特别提到,逆合成这类工具明明早就有了,但大多数小实验室的化学家从来不用。门槛——授权、专门训练、专用软件的搭建——本身就是一种障碍。而Claude把这些能力变成了将数据粘贴进对话框这样低的使用成本,这件事对那些一直被专业工具挡在门外的人,可能比追平精度更有意义。
最后是它的姿态。这是一篇由化学家主笔、把局限一条条摊开、反复强调补充而非替代化学家判断的研究。它没有喊口号,它是在做基准、认偏差、划边界。对于一个本质上还很难被验证的领域,这种克制和诚实,本身就是可信度的一部分。
顺带一提,Anthropic也在将其AI for Science计划更明确地扩展到化学研究,并公开征集那些Claude可能帮上忙、尤其涉及这类多模态推理的课题。换句话说,这篇白皮书与其说是一个终点,不如说是一份邀请函。
参考文献
Anthropic 研究博客《Making Claude a chemist》及配套白皮书,作者 Da vid Kamber。
本文为基于该原文的中文深度解读,具体数据与结论以原文为准。
https://www.anthropic.com/research/making-claude-a-chemist





