处理非结构化数据一直是个挑战——无论是社交网络里的错综关系,还是化学分子里的原子连接。传统的深度学习模型,像处理图像的卷积网络或者处理文本的循环网络,面对这种“不规则”数据结构时往往力不从心。而图神经网络,正是为这类图结构数据量身打造的深度学习方法。
简单来说,GNN 的核心在于“信息传递”。它把图里的每个节点看作一个实体,通过反复聚合邻居节点的信息,来不断更新自己对当前节点的理解。这种方式让模型能够高效地学习图中复杂的拓扑关系和节点属性,从预测分子性质到推荐系统,应用前景相当广阔。

分子的图结构
理论说完了,咱们来点实际的。就拿一个最简单的分子——乙醇(化学式 C₂H₆O)来举例,一步步看它如何变成一个GNN能理解的图数据结构。
要理解GNN,首先要明白它的输入不再是规整的像素矩阵或者一维序列,而是一个实实在在的图。直接把它用在分子上,第一步当然是把乙醇分子抽象成一个数学意义上的图。
节点与邻接关系
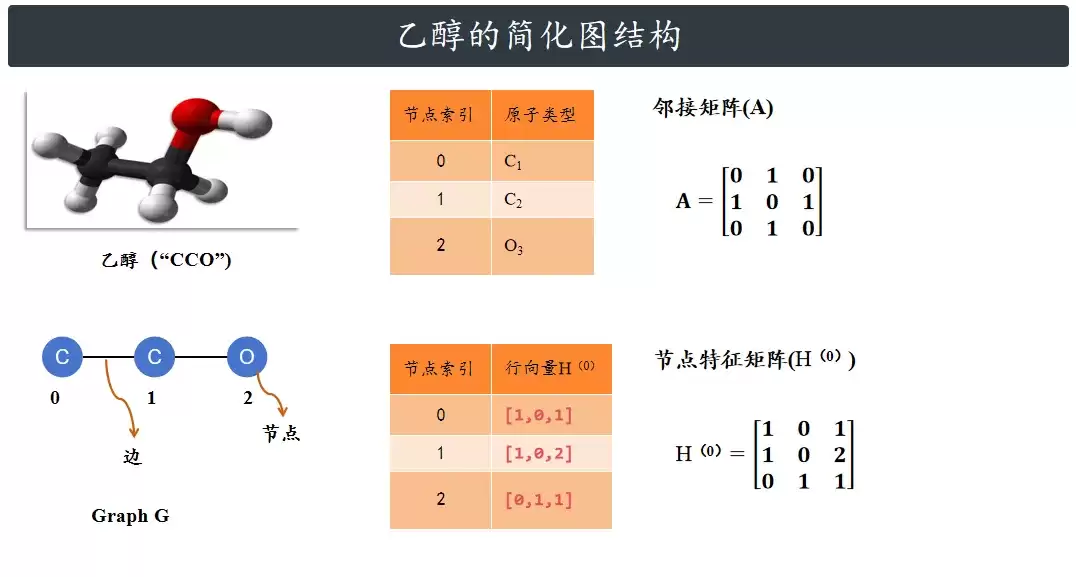
为了演示起来更直观,这里先忽略氢原子,只盯着三个重原子看:两个碳原子(C)和一个氧原子(O)。氢原子的影响我们通过节点的特征来间接体现。
那么,怎么描述这些原子之间的连接关系呢?靠邻接矩阵。如果两个原子之间有化学键,矩阵中对应的位置就是1,否则是0。
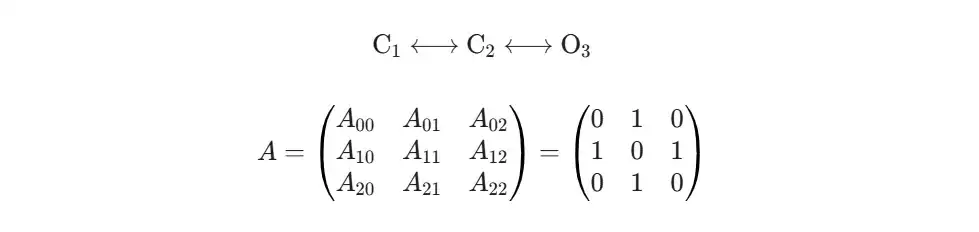
下一步,是给每个原子赋予一些初始属性,这就是节点特征矩阵。为了演示,我们设计一个简单的特征向量,比如用独热编码区分原子种类,再加上它的连接度(跟几个原子相连)。这样,每个节点就有了一个数字化的“身份”。这就是GCN模型真正的输入数据。
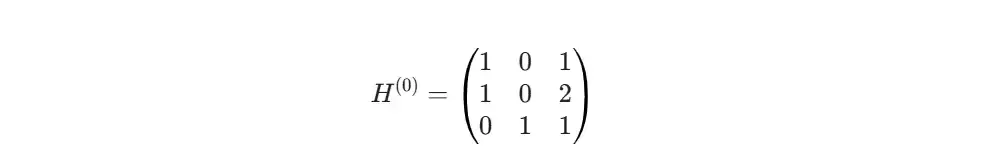
当然,真实的分子图项目远比这个复杂。虽然用到的“节点”和“边”的基本原理一样,但节点特征和图的结构复杂性会成倍增加。在PyTorch Geometric(PyG)或Deep Graph Library(DGL)这样的专业图神经网络库中,借助像RDKit这样的化学信息学工具,分子图的定义会详尽得多。关于这部分,后面我们会专门展开聊。
图卷积
数据和结构都准备好了,核心的“图卷积”操作就可以登场了。它的输入就是前面得到的节点特征矩阵 H 和邻接矩阵 A。我们用一个3个节点、每个节点特征维度为3的小例子来演示。图卷积的计算可以拆解为两个关键步骤:节点特征的线性变换 和 邻接特征的聚合。
GCN 单层的核心公式是:
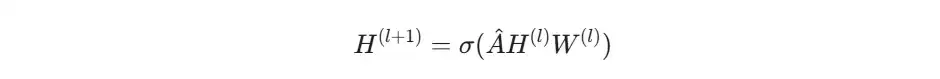
1. 节点特征的线性变换
这一步非常直观,跟普通神经网络里全连接层的思路一样:用一个可学习的权重矩阵 W 对原始特征 H 做一次线性变换,提取更高层的特征,同时改变特征的维度。经过这一步,我们就得到了每个原子经过权重变换后的新特征 X'。
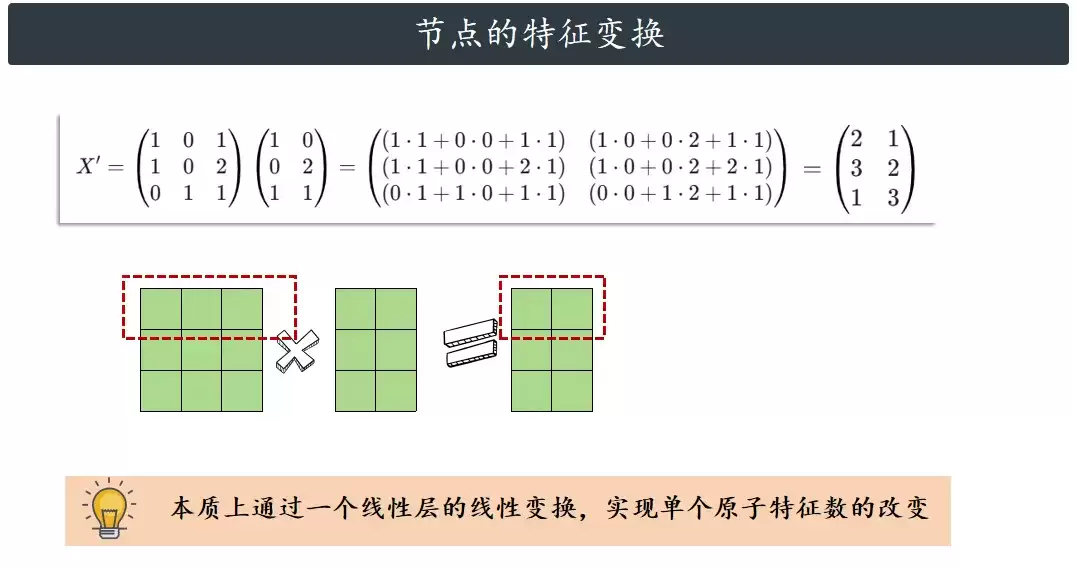
2. 邻接特征的聚合
这才是图卷积的精髓所在。为了方便理解,我们直接用邻接矩阵 A 来进行聚合操作。这个操作的本质,就是把每个节点所有邻居的特征信息“拉”到自己身上来。具体怎么“拉”?就是通过邻接矩阵 A 与变换后的特征 X' 相乘。矩阵 A 中每个元素的值,定义了消息如何从邻居节点传递过来并加权求和。


到这里,一次图卷积就完成了。这个新特征矩阵,就是聚合了邻居信息之后再经过特征变换的结果。总结来看,一次图卷积,本质上是一个“先变换,再聚合”的过程。在这个过程中,节点特征矩阵是不断被更新的“数据”,而邻接矩阵定义了图的结构和消息传递路径,是相对固定的。
分子图的表达方式
之前为了演示,把问题简化了。在真实的项目中,分子图的构建远不止这么简单。一个标准的流程是这样的:从文本化的分子表达(SMILES字符串)开始,经过RDKit这样的专业化学库解析,最后转化为PyG或DGL能够处理的图对象。
真实项目中分子表示的流程:
SMILES 字符串 → RDKit 解析 → PyG/DGL 图对象
先把需要的依赖装上:
pip install rdkit
pip install torch_geometric
# 假设 PyTorch, RDKit, PyTorch Geometric (PyG) 库已安装 import torch from rdkit import Chem from torch_geometric.utils.smiles import from_smiles # PyG中用于SMILES转换的实用函数 (或使用更早版本的'from_rdkit') # --- 1. 定义和转换 --- smiles_ethanol = "CCO" # 使用 PyG 的封装函数,一步完成解析、特征提取和图结构构建 # 这个函数内部自动完成了原子特征编码、键索引构建、以及 PyTorch 张量转换。 ethanol_data = from_smiles(smiles_ethanol) # --- 2. 展示结果 --- print("=" * 40) print(f"乙醇分子 SMILES: {smiles_ethanol}") print("PyG Data 对象结构 (封装结果)") print("=" * 40) # PyG Data 对象概览 print(ethanol_data) print("n--- 关键张量尺寸分析 ---") print(f"节点特征矩阵 (x): {ethanol_data.x.shape}") print(f"邻接信息 (edge_index): {ethanol_data.edge_index.shape}") print("-" * 40)
运行代码后,会输出类似下面的结果:
========================================
乙醇分子 SMILES: CCO
PyG Data 对象结构 (封装结果)
========================================
Data(x=[3, 9], edge_index=[2, 4], edge_attr=[4, 3], smiles='CCO')
--- 关键张量尺寸分析 ---
节点特征矩阵 (x): torch.Size([3, 9])
邻接信息 (edge_index): torch.Size([2, 4])
----------------------------------------
PyG Data 对象结构解读
这里需要解释一下输出中的维度含义。为什么是3个节点?因为PyG的默认处理方式,是只把重原子看作节点,忽略氢原子,所以乙醇(CCO)就是3个节点。9(列)代表每个节点有9个特征维度,这些特征是`from_smiles`函数内部自动计算出的化学属性,比如原子类型、价态、电荷等。
对于边,PyG使用`edge_index`(2行)来表示图结构。第一行是源节点索引,第二行是目标节点索引。4列代表有4条有向边(因为化学键默认是双向的,2个C-C单键 + 2个C-O单键,每条键都产生两条有向边)。
图卷积神经网络
有了前面的基础,现在终于可以把真实数据塞进一个真正的图卷积神经网络里进行前向传播了。我们继续用乙醇分子作为例子,模拟这个完整的过程。
直观地看,张量的变换过程是这样的:
- 原始输入是一个大小为 3×9 的张量(3个节点,每个节点9个特征)。
- 经过第一个隐藏层通道数为16的图卷积,我们得到一个 3×16 的张量。
- 再经过第二个隐藏层通道数为16的图卷积,同样得到一个 3×16 的张量。
- 接着是一个全局平均池化层。顾名思义,就是把所有节点在同一个特征维度上取平均值,把整个图的特征浓缩成一个向量,得到一个 1×16 的图级特征。
- 最后,通过一个线性分类层,将这个 1×16 的特征转化为最终的预测结果,比如一个表示分子某种性质的数值。
代码实现
下面是用PyTorch Geometric实现这个过程的完整代码:
import torch import torch.nn.functional as F from torch_geometric.nn import GCNConv, global_mean_pool from torch_geometric.data import Data import sys # 尝试导入 RDKit 和 PyG 转换工具 from rdkit import Chem from torch_geometric.utils.smiles import from_smiles # --- 1. 真实数据准备 (使用 PyG 封装函数) --- smiles_ethanol = "CCO" # 一步转换:生成包含所有 9 个原子(C, C, O, 6H)的图结构 ethanol_data = from_smiles(smiles_ethanol) # 修复:确保节点特征是浮点类型 (解决 RuntimeError) ethanol_data.x = ethanol_data.x.float() # 生成 Batch Tensor:9 个节点都属于同一个图 (batch size=1) N_NODES = ethanol_data.x.shape[0] ethanol_data.batch = torch.zeros(N_NODES, dtype=torch.long) # --- 2. 展示输入数据结构 --- print("=" * 60) print(f"乙醇分子 SMILES: {smiles_ethanol}") print("PyG Data 对象结构 (GCN 模型输入 - 真实配置)") print("=" * 60) print(f"节点数 N: {ethanol_data.x.shape[0]}") print(f"节点特征矩阵 (x): {ethanol_data.x.shape}") print(f"邻接信息 (edge_index): {ethanol_data.edge_index.shape}") print("-" * 60) # --- 3. 定义 GCN 模型类 (SimpleGCN) --- class SimpleGCN(torch.nn.Module): def __init__(self, num_node_features, hidden_channels, num_classes): super().__init__() self.conv1 = GCNConv(num_node_features, hidden_channels) self.conv2 = GCNConv(hidden_channels, hidden_channels) self.lin = torch.nn.Linear(hidden_channels, num_classes) def forward(self, x, edge_index, batch): print(f"n[A] 初始输入 x (H(0)): {x.shape}") # GCN 层 1 x = self.conv1(x, edge_index) print(f"[B] GCNConv 1 输出 (H(1)): {x.shape}") x = F.relu(x) # GCN 层 2 x = self.conv2(x, edge_index) print(f"[C] GCNConv 2 输出 (H(2)): {x.shape}") x = F.relu(x) # 全局读出/池化层 x = global_mean_pool(x, batch) print(f"[D] Global Mean Pool 输出: {x.shape} <--- **图级特征**") # 线性分类层 x = self.lin(x) print(f"[E] 最终分类层输出: {x.shape} <--- **预测结果**") return x # --- 4. 模型实例化与运行 --- # 定义模型参数 INPUT_DIM = ethanol_data.x.shape[1] # 自动获取真实/模拟的特征维度 (例如:11) HIDDEN_DIM = 16 OUTPUT_DIM = 1 # 实例化模型 model = SimpleGCN( num_node_features=INPUT_DIM, hidden_channels=HIDDEN_DIM, num_classes=OUTPUT_DIM ) # 执行前向传播 print("=" * 60) print(f"【Simple GCN 前向传播过程(隐藏层维度 D_hidden={HIDDEN_DIM})】") print("=" * 60) output = model( ethanol_data.x, ethanol_data.edge_index, ethanol_data.batch )
运行上述代码,终端会清晰地打印出每一步张量的形状变化:
============================================================
乙醇分子 SMILES: CCO
PyG Data 对象结构 (GCN 模型输入 - 真实配置)
============================================================
节点数 N: 3
节点特征矩阵 (x): torch.Size([3, 9])
邻接信息 (edge_index): torch.Size([2, 4])
------------------------------------------------------------
============================================================
【Simple GCN 前向传播过程(隐藏层维度 D_hidden=16)】
============================================================
[A] 初始输入 x (H(0)): torch.Size([3, 9])
[B] GCNConv 1 输出 (H(1)): torch.Size([3, 16])
[C] GCNConv 2 输出 (H(2)): torch.Size([3, 16])
[D] Global Mean Pool 输出: torch.Size([1, 16]) <--- **图级特征**
[E] 最终分类层输出: torch.Size([1, 1]) <--- **预测结果**
整个过程非常直观:输入3个节点、9维特征,经过两层GCN卷积,特征维度升到16维,全局池化把3个节点压缩成一个图级别的16维向量,最后线性层输出一个单一的预测值。这就是一个分子图分类的最基础流程了。