计算机辅助药物设计流程梳理
本文系统梳理分子对接与分子动力学的核心内容,全面回顾计算机辅助药物设计的完整流程。小分子药物研发是发展迅猛的前沿领域,有望彻底改变新药开发的模式与效率。随着结构生物学和计算方法的持续突破,研究人员对分子结构及药效关系有了更深刻的认识,正致力于设计出比以往更高效、更具选择性、更安全的小分子化合物,并不断探索创新方案。
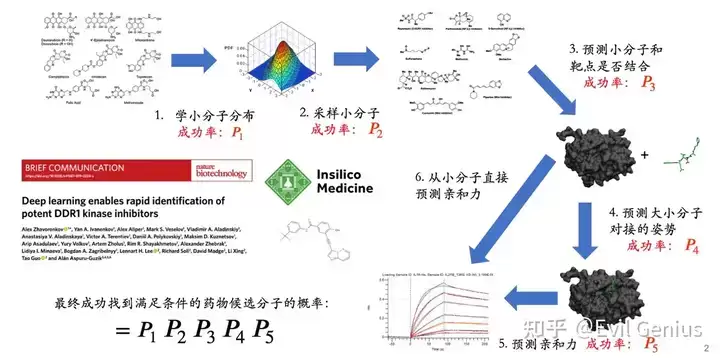
第一步:预测蛋白质三维结构
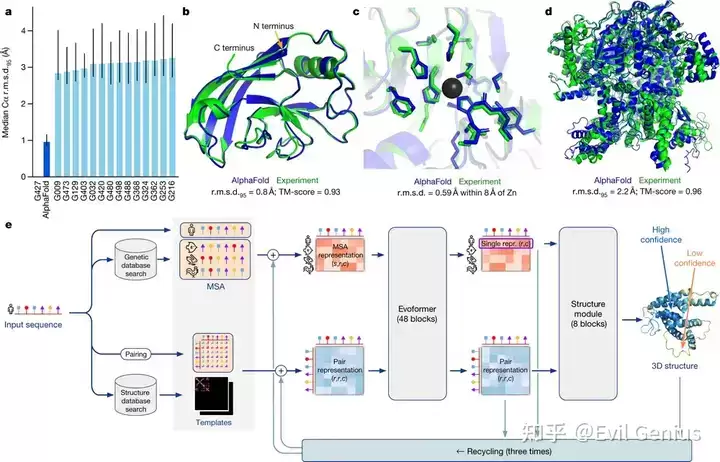
蛋白质三维结构的获取方式多样,包括X射线衍射、冷冻电镜、核磁共振波谱以及计算预测技术。准确预测三维结构已成为药物设计的关键环节,近年来备受关注。深入理解蛋白质的三维构象,有助于阐明药物结合机制,促进设计亲和力更强、特异性更高的候选化合物。值得注意的是,计算技术的飞速发展已为药物发现领域带来革命性变化,蛋白质结构预测的精度也达到了前所未有的水平。
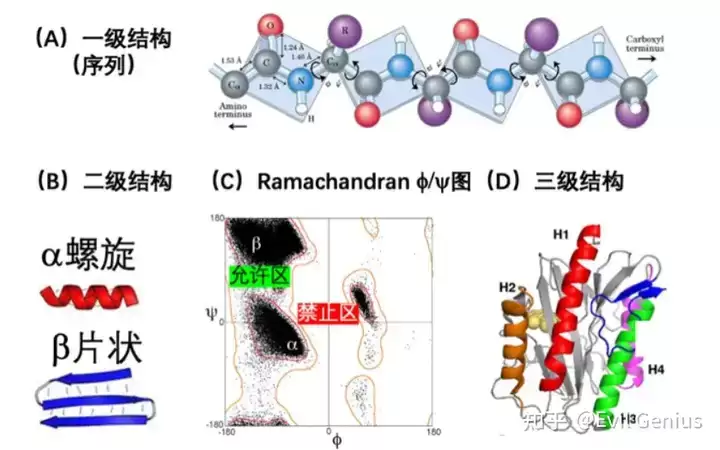
在预测蛋白质结构的诸多计算方法中,同源建模应用最为广泛。该技术又称比较建模,利用已知相关蛋白质的结构推测目标蛋白质的结构。其基本假设是:进化上具有亲缘关系的蛋白质通常拥有相似的结构,且结构保守性高于序列保守性。
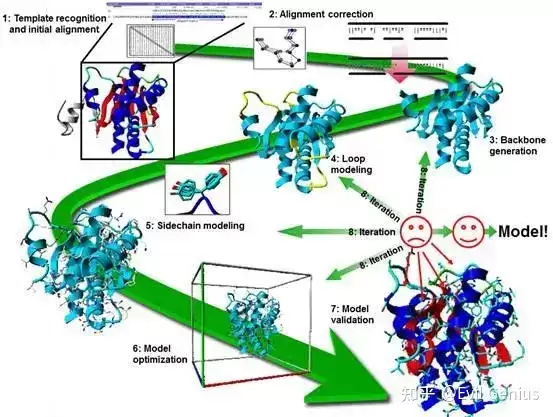
具体流程包括:
- 模板选择:从蛋白质结构数据库中挑选合适的模板结构。模板需与目标蛋白质序列具有较高的同源性,并具备相似的功能或折叠类型。常用工具如BLAST用于识别同源序列。
- 序列比对:将目标蛋白质序列与所选模板结构进行比对,优化对齐效果,确保保守区域得到正确匹配。常用算法包括ClustalW和MUSCLE。
- 模型构建:基于比对结果生成目标蛋白质的三维模型——将目标序列叠合到模板结构上,调整侧链以适配目标序列。常用软件有MODELLER和SWISS-MODEL。
- 模型优化:初始模型生成后需进一步优化以提升准确性,包括调整侧链取向、进行能量最小化以及结构验证,确保模型在生物学上合理。常用软件如CHARMM和GROMACS。
另一种方法是从头预测(de novo),该方法不依赖已知结构,完全基于物理原理(如能量最小化和自由能计算)来预测蛋白质结构。
- 片段组装:将蛋白质序列拆分为小片段,预测这些片段在三维空间中的相对取向。可采用物理法(计算系统能量)或统计法(基于已知结构的片段组装概率)。
- 模型构建:以满足空间约束的方式连接各片段,构建完整的三维模型。该过程由基于知识的能量函数或机器学习算法引导。
- 模型优化:通过分子动力学模拟、能量最小化或机器学习方法对模型进行优化,提高精度和质量。


同源建模、从头预测等计算方法使蛋白质结构预测达到高精度,为药物发现带来了深刻变革。准确预测蛋白质结构的能力,还推动了针对过去被认为“不可成药”靶点的新药开发。
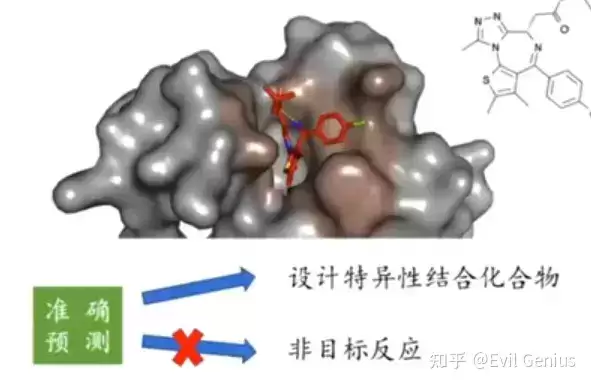
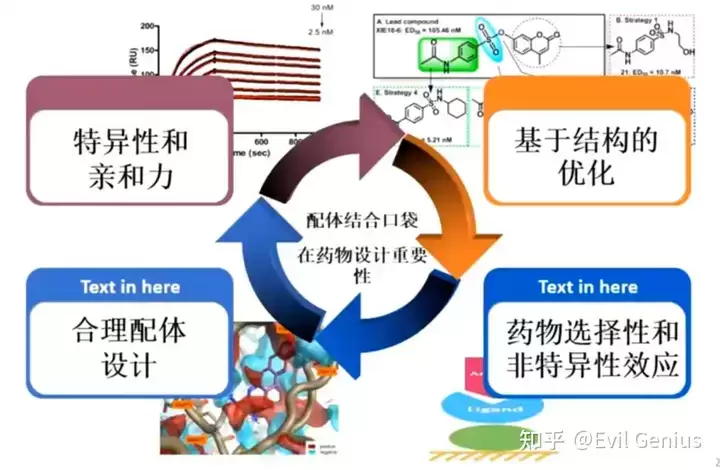
配体结合口袋在SBDD中的关键作用
配体结合口袋在基于结构的小分子药物设计中扮演核心角色。设计思路的起点在于:药物分子通过与靶向蛋白质(通常为蛋白质)发生特异性结合,从而发挥生物活性。
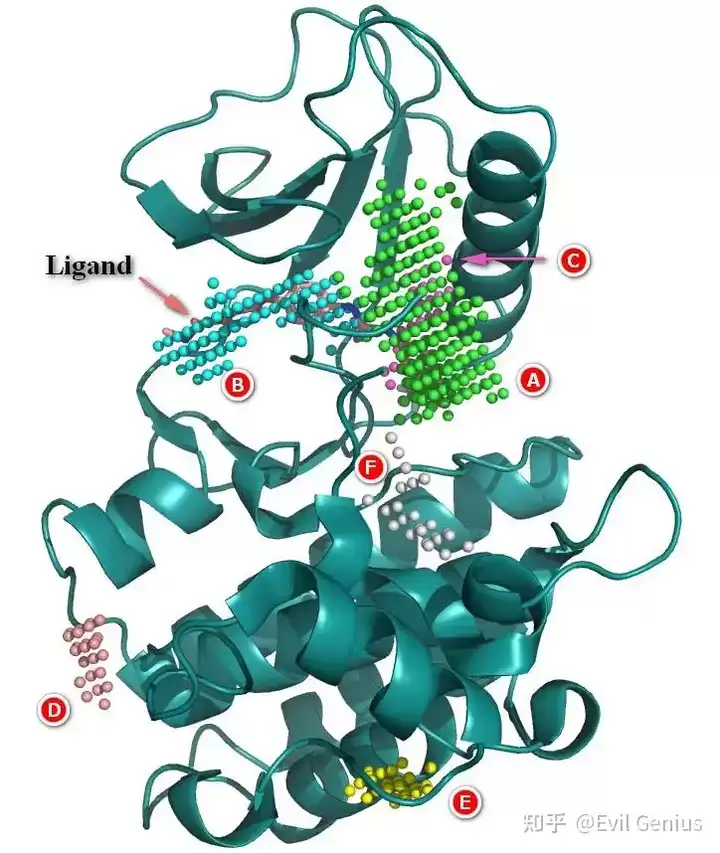
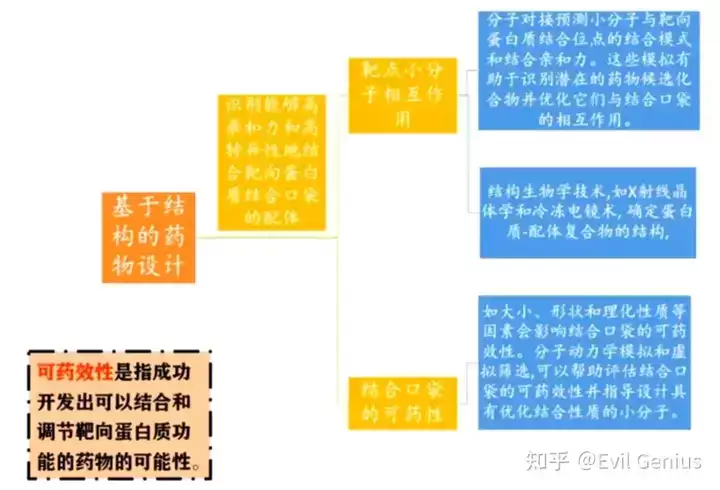
第二步:小分子药物的结构分析
药物主要分为小分子药物和生物制剂两大类。小分子药物通常通过化学合成获得,结构相对简单,设计目标是与疾病相关的特定分子实现高特异性结合。生物制剂则是由活细胞产生的大型复杂分子(如蛋白质),通过与体内特定受体或通路相互作用发挥功效。
官能团是小分子药物设计的关键结构特征之一。例如羟基(-OH)、羧基(-COOH)、氨基(-NH₂)等官能团的存在,可使小分子与特定酶或受体相互作用,并在体内赋予特定的药理学特性。
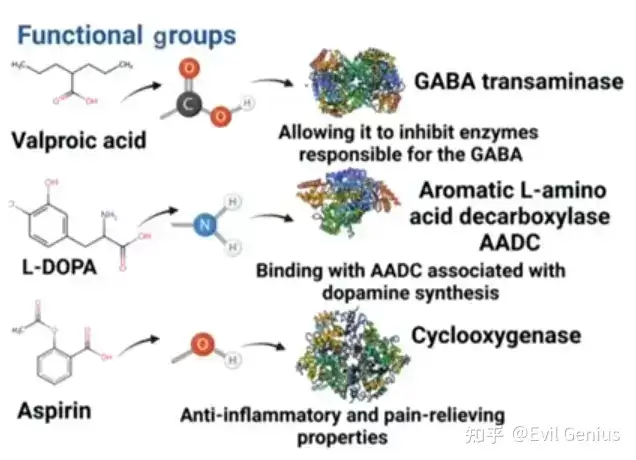
立体化学是小分子药物设计的另一个重要维度,指分子中原子的三维排布如何影响其与生物靶点的相互作用。例如抗炎药异布比酮的活性形式具有特定的立体化学构型,使其能够与环加氧酶结合并抑制其活性。
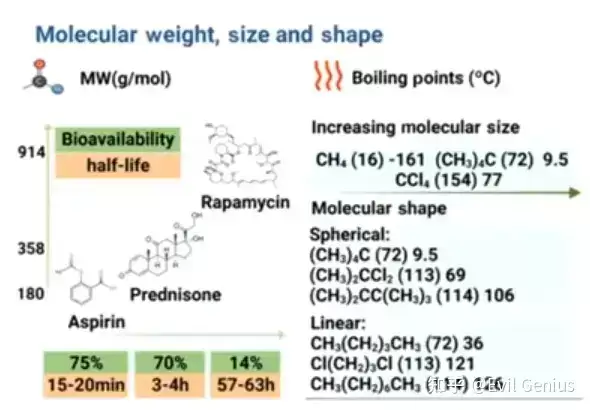
分子量同样不容忽视,它对药代动力学和药效动力学均有影响。低分子量药物通常吸收和排泄较快,而高分子量药物可能具有更长的半衰期、所需剂量更低。分子的大小和形状会影响其物理化学性质,例如较大分子往往具有更高的沸点和熔点。
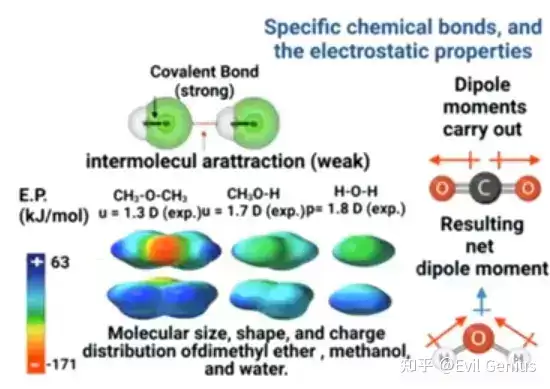
分子中特定化学键的存在决定了其结构与性质。共价键通过两个原子之间共享价壳电子对形成,IV族至VI族的原子通过结合来填满价壳层的八电子稳定态。分子的电荷特性(如偶极矩和电荷分布)影响其与其他分子的相互作用及反应活性。理解这些结构特征,研究人员便能设计出药理学特性更优、副作用更少的小分子候选化合物。
第三步:小分子药物设计的计算方法
分子对接
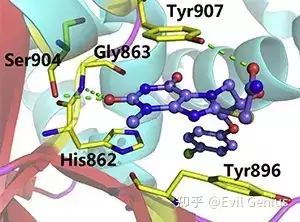
分子对接是小分子药物设计中应用最广泛的计算方法之一。该技术通过预测小分子(配体)与较大靶点(如蛋白质或核酸)之间的结合亲和力与取向,为药物研发提供关键信息。对接算法利用能量最小化和评分函数来评估配体-蛋白质复合物的稳定性,并对潜在候选化合物进行排序。通过对接过程,研究人员能够深入理解多种生物过程(如药物-蛋白质相互作用、蛋白质-蛋白质相互作用及酶促反应)的分子机制。随着计算机技术与生命科学的深度融合,分子对接的精确性和效率已显著提升。
具体进展包括:
- 自由能微扰(FEP)和热力学积分(TI)等计算方法被开发出来,可提供比传统对接更准确的结合亲和力预测。
- 新型对接工具如HADDOCK和RosettaDock引入了蛋白质柔性,能够预测配体结合所引发的构象变化,从而提升预测准确性。
- 机器学习被整合进对接工具中,例如用随机森林回归替代传统线性回归方法,显著改善了预测性能。通过训练已知配体-蛋白质复合物(带有实验测定的结合亲和力数据),机器学习模型可学习模式并对新的未知复合物进行预测。
虚拟筛选
虚拟筛选借助分子对接或其他技术,对大型化合物库进行高效筛选,以识别潜在的药物候选分子。该策略能够大幅节省成本与时间,帮助研究人员缩小需要进一步实验验证的化合物范围。常用工具包括GOLD、GLIDE以及AutoDock Vina(免费且效果较佳的选择)。
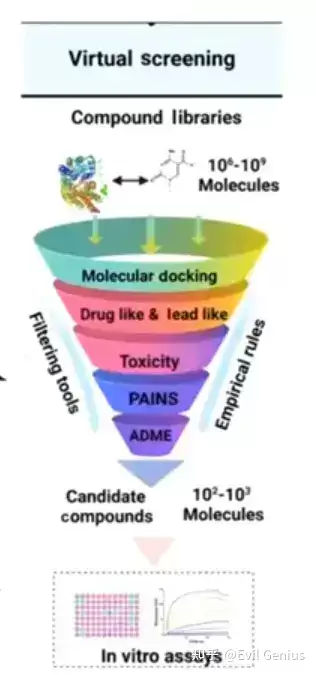
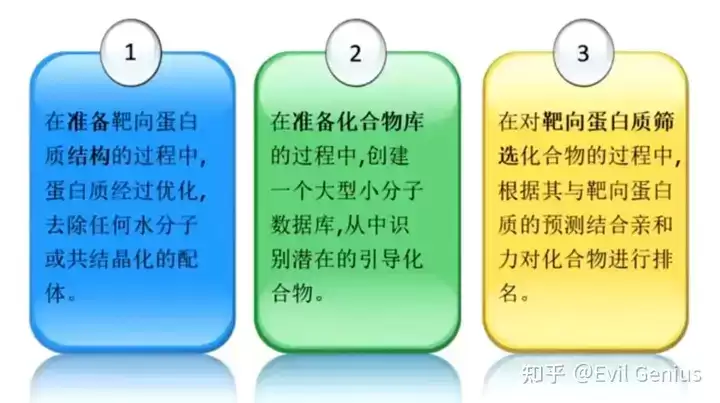
需要指出的是,虚拟筛选结果的准确性高度依赖于蛋白质结构与化合物库的质量。筛选过程通常未考虑药代动力学和毒理学特性,因此通过虚拟筛选识别的化合物仍需实验验证,以确认其疗效与安全性。
分子动力学模拟
分子动力学(MD)模拟是一种强大的计算工具,用于研究体系中原子与分子的物理运动及相互作用。通过对牛顿运动方程进行时间积分,可获得原子的运动轨迹,从而在原子层面提供系统宏观行为的定量与定性信息。简而言之,MD模拟犹如一部记录系统动态演化的“电影”。

近年来的进展:
- 深度学习算法(如原子卷积网络)可直接根据原子坐标预测分子势能,显著提升了模拟精度。
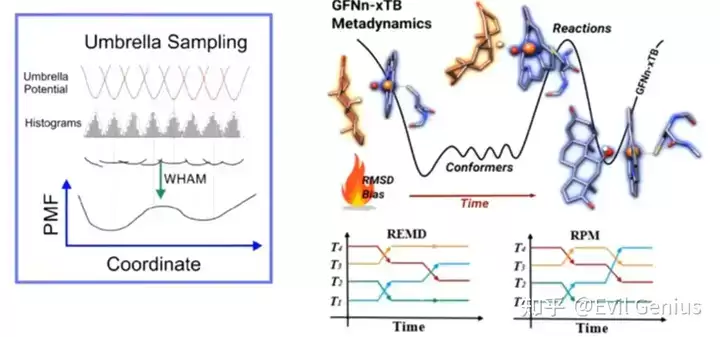
- 增强采样技术(如metadynamics)克服了传统MD的局限性,能够探索更大的构象空间,更准确地反映系统能量面。
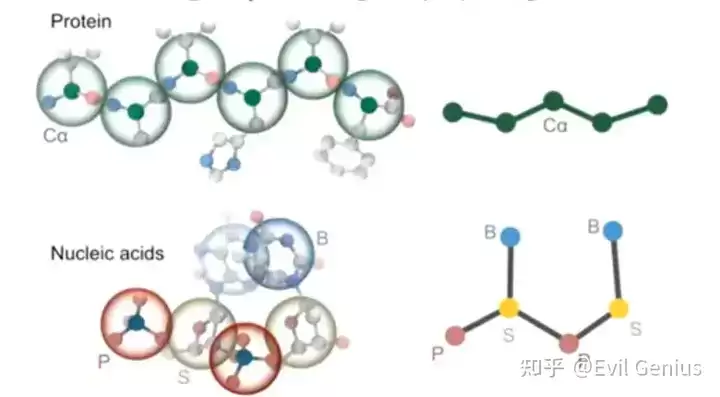
- 粗粒度模型通过将原子分组简化系统表示,降低计算成本,从而能够模拟传统全原子模型无法承受的更大体系与更长时间尺度。
量子力学/分子力学计算
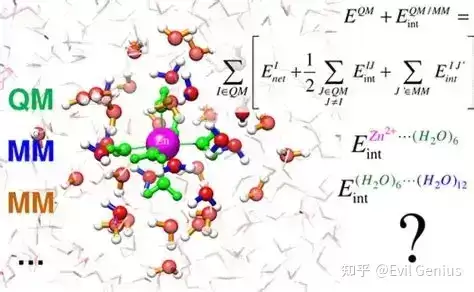
QM/MM计算将量子力学的精确性与分子力学的速度相结合,用于研究化学与生化系统。该方法将QM区域视为溶质(或关键区域),MM区域视为溶剂(或非关键区域),从而能够考虑溶剂化效应。该技术由Warshel和Levitt于1976年首次提出,现已广泛应用于酶催化机制、反应路径及蛋白质-配体结合相互作用的研究。与传统分子力学或量子力学仅考虑固定构型不同,QM/MM能够提供准确的反应能与反应路径计算,并处理仅依靠量子力学难以高效模拟的大型系统。
机器学习
机器学习算法在预测小分子性质方面日益受到青睐,涵盖溶解度、毒性、结合亲和力等指标。这些算法有望彻底革新药物发现模式,减少对昂贵实验方法的依赖,使药物开发更加快速高效。
当前计算方法的局限性
尽管前景广阔,但现有方法仍面临若干关键挑战:
- 蛋白质柔性:传统对接通常将蛋白质视为刚性体,这种简化过于理想化。蛋白质具有高度动态性,考虑其柔性是一个巨大难题。
- 评分函数:用于预测结合亲和力的评分函数往往不够精确,基于简化模型,难以完全捕捉相互作用的复杂性。
- 溶剂效应:许多对接算法未充分考虑溶剂的作用,而溶剂在分子相互作用中至关重要,忽略它会导致预测偏差。
- 时间尺度:MD模拟受限于可模拟的时间范围(纳秒至微秒),而众多生物过程发生在毫秒乃至秒级尺度。
- 计算成本:对接和MD模拟均计算量巨大,限制了可研究的系统规模与模拟时长。
- 实验数据匮乏:许多预测缺乏实验数据验证,难以评估其准确性。
- 诱导契合预测困难:大量算法难以预测配体结合时蛋白质的构象变化。
- 非靶向效应预测局限:虽然对预期靶点的相互作用预测能力在提升,但预测非靶向效应仍是重大挑战。
以上就是计算机辅助药物设计从蛋白质结构预测到计算方法的核心流程。限于篇幅,许多细节无法一一展开,但整体框架有助于快速建立系统认知。




