先说几个核心判断:AlphaFold2确是一场变革,但其应用边界同样清晰。在基于结构的药物设计(SBDD)中,AF2输出的是单一最优静态结构,构象多样性方面存在天然缺陷。对于GPCR这类具有高度构象动态性的重要药物靶标,这一问题尤为突出。近期一项名为AFsample2T的研究提出了一种富有启发性的解决方案——在不修改AF2网络架构的前提下,通过在MSA输入层面施加局部"遮蔽",精准释放结合口袋区域的进化约束,从而生成更具多样性的构象集成。
研究背景
AF2的里程碑意义与内在局限
AlphaFold2在CASP14上的表现毋庸置疑——它以近乎碾压的方式超越了所有传统方法。然而,当将其真正应用于基于结构的药物设计时,问题逐渐显现:AF2的训练目标决定了它只会输出那个"最正确"的静态结构。其Evoformer模块通过行向与列向注意力机制,从多序列比对(MSA)中充分挖掘共进化信号,然后将这些信号牢牢"锁定"在预测结构中。结果呢?构象多样性几乎为零。
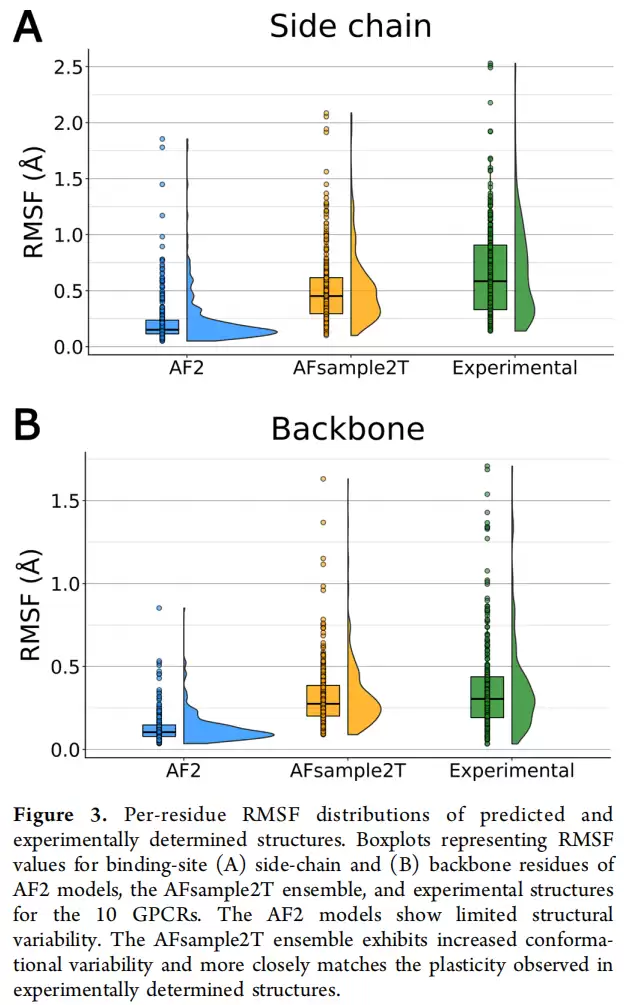
具体数字很有说服力:结合口袋侧链的RMSF中位值仅为0.15Å,而实验结构集合的RMSF高达0.58Å;骨架RMSF中位值为0.10Å,实验结构则是0.30Å。更直观的是,AF2生成的口袋普遍偏"塌陷"、偏小,平均体积约209ų,而实验结构的均值约为256ų。这就好比用一张照片去捕捉一段舞蹈——定格固然清晰,但动态信息全都丢失了。
GPCR:最重要也最棘手的药物靶标
G蛋白偶联受体(GPCR)是人类最大的膜蛋白超家族,约三分之一的上市药物以其为靶点。但GPCR的结构特征决定了它对构象采样极其敏感:正构结合位点位于跨膜螺旋TM1-7的胞外侧,由于内源性配体包括肽类、小分子、脂质等,口袋形态高度多样;受体本身存在激活态与非激活态两大功能构象,TM6的向外位移是关键标志;再加上胞外环2(EL2)在不同配体复合物中的构象异质性——这种天然的构象多样性,AF2的默认策略根本难以捕捉。多项回顾性评估已明确指出,AF2模型在GPCR虚拟筛选中的表现系统性差于实验结构,根源就在于结合口袋的精细描述不足。
方法设计:AFsample2T的核心策略
MSA列蒙版的原理
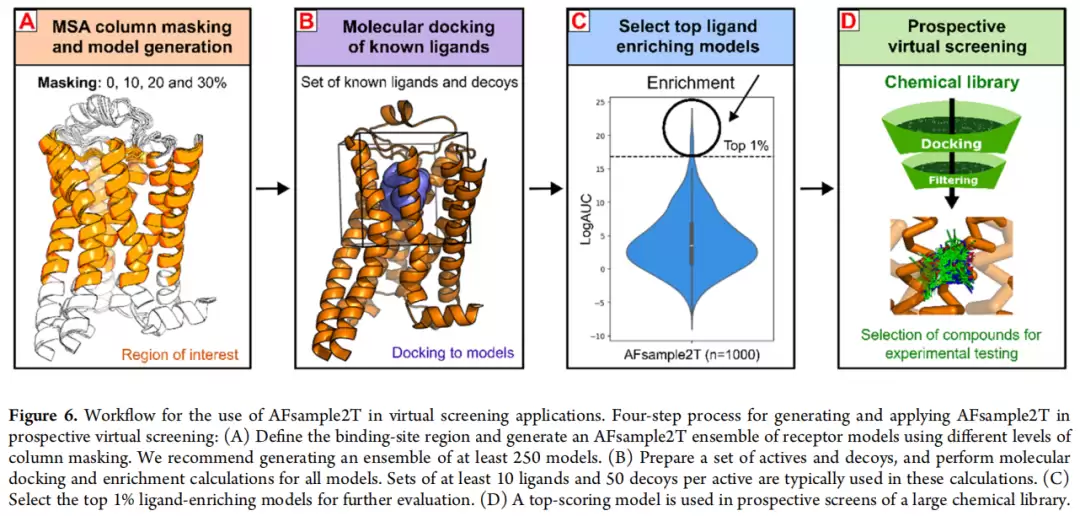
所谓MSA列蒙版,逻辑其实很直观:通过随机遮蔽MSA中特定位置的列,削减Evoformer可获取的共进化信息量,让AF2在结构推断时"不得不"走出原有的舒适区,探索偏离进化约束的构象空间,进而产生更高的结构多样性。已有工作(AFsample2,Kalakoti & Wallner, Commun. Biol. 2025)表明,对全MSA施加15%蒙版可增加整体构象多样性,而本篇工作的创新在于将蒙版精确靶向至结合口袋区域。
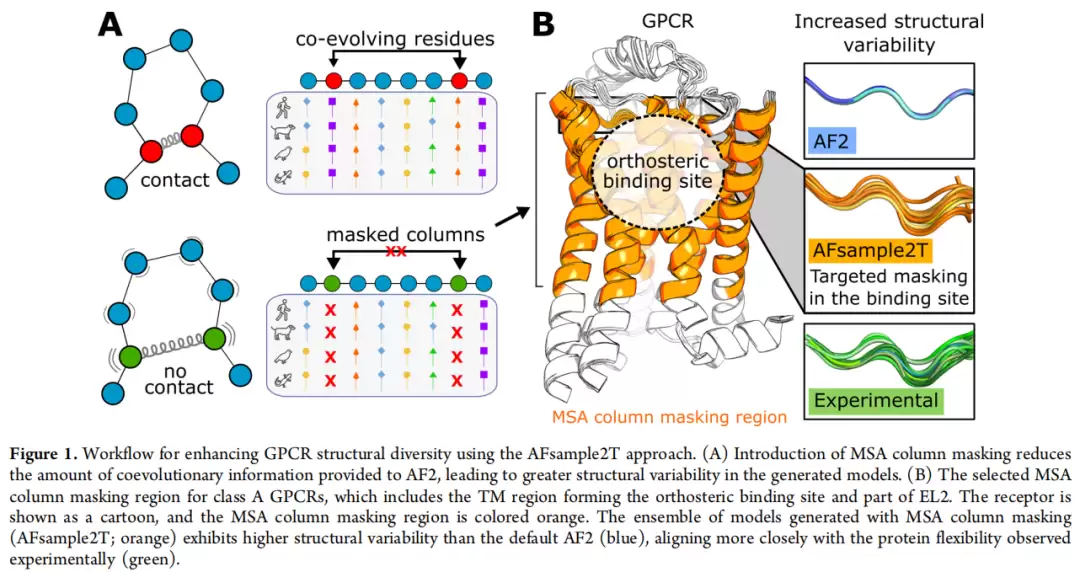
AFsample2T的靶向设计
蒙版区域定义(针对class A GPCR通用):
| 区域 | 具体范围 | 作用 |
|---|---|---|
| 胞外TM区 | TM螺旋胞外侧,延伸至PIF保守微开关(P5×50, I3×40, F6×44)以上一个螺旋圈 | 覆盖正构口袋的主体 |
| EL2区 | 保守半胱氨酸至TM5之间的EL2片段 | 捕获环区构象异质性 |
蒙版概率梯度测试了0%(仅dropout)、10%、20%、30%、50%五档。
集成策略与激活/非激活态建模
AFsample2T的集成组成如下:每个受体生成1,000个模型,其中0%蒙版加dropout 250个(激活态和非激活态各125个),10%蒙版加dropout 250个,20%蒙版加dropout 250个,30%蒙版加dropout 250个。
两大功能构象的建模也颇具匠心——非激活态仅输入受体序列(AF2 monomer),激活态则输入受体序列加上异三聚体G蛋白(Gα/β/γ)序列(AF2-Multimer),通过G蛋白的胞内结合来引导TM6向外位移。验证表明,这套策略可以可靠地区分TM6的特征性构象,与Chiesa et al. 2025的结果一致。
实验设计与评估体系
结构精度评估(10个class A GPCR)
基准数据集涵盖了10个GPCR受体,包括5-HT₁A、A₂A、D₁、D₂、H₁、M₄、MT₁、μ-阿片受体、TAAR1等,共119个实验结构,内源性配体类型从单胺、嘌呤核苷到内源性肽,相当多样化。排除低分辨率结构、重复配体复合物和结合位点突变体后,保留了61个结构用于精度基准测试。
评估指标方面,以结合位点残基(配体周围5Å内)的对称感知侧链RMSD为核心指标,计算不同RMSD阈值(1.0-2.0Å)下实验结构被捕获的比例曲线并用AUC量化;同时使用pLDDT和PAE评估模型置信度,使用MDTraj计算RMSF评估集成构象多样性,使用Schrödinger SiteMap计算口袋体积。
虚拟筛选评估
分子对接工具选用DOCK3.8,基于物理评分函数,包含vdW、静电、配体去溶剂化项。配体集构建方面,活性化合物从ChEMBL v33获取(pKᵢ/pKd/pIC₅₀/pEC₅₀ ≥ 6.0),经标准化和聚类后,每受体保留52-202个配体;诱饵分子从ZINC20按性质匹配生成,每受体2,580-10,375个。整体评估规模相当庞大——对10个GPCR的所有模型(AF2加AFsample2T各1,000个)及119个实验结构进行对接,累计预测打分超过240万亿个复合物构象。评估指标包括LogAUC / aLogAUC(ROC曲线半对数面积)和EF1%(ROC曲线在1%假阳性率处的早期富集因子),分析维度覆盖集成的中位值和top 1%最大值。
核心结果
构象精度:全面优于默认AF2
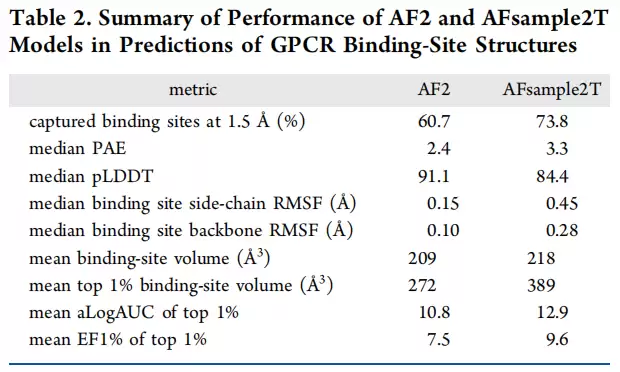
结合位点RMSD-AUC的汇总数据相当有说服力:默认AF2的AUC为0.54,仅添加dropout(0%蒙版)就能提升到0.57;10%和20%靶向蒙版均达到0.61,相对AF2提升约13%;30%蒙版为0.59,小幅下降;50%蒙版则降至0.43,骨架已被破坏。值得注意的是,全局15%蒙版的AFsample2反而只有0.38,低于AF2基线。而AFsample2T集成(0-30%蒙版)表现最优,AUC达0.63,相对AF2提升17%。在RMSD ≤1.5Å阈值下,AFsample2T集成捕获了73.8%的实验结合位点构象,AF2仅为60.7%,提升22%。
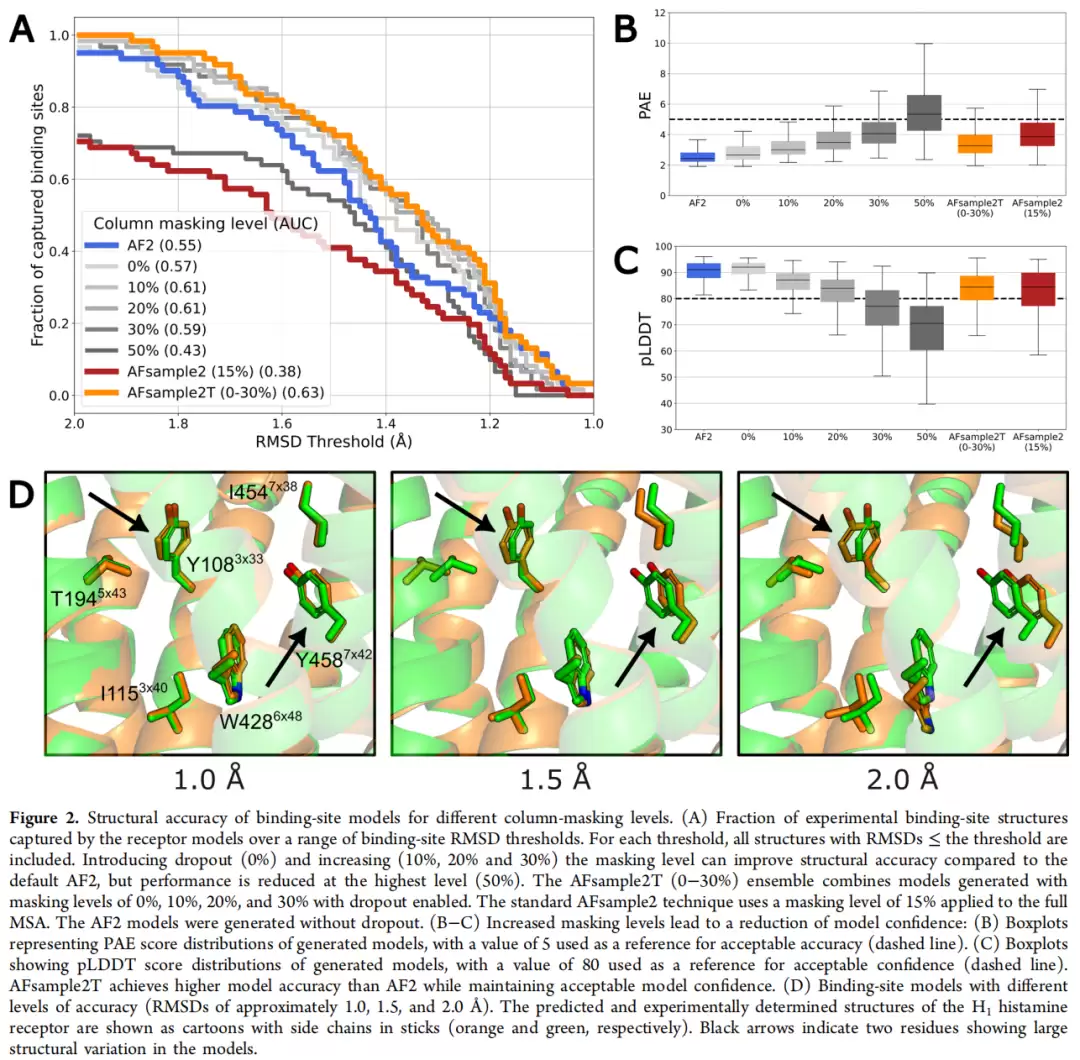
构象多样性:接近实验水平
RMSF对比数据更直观地展示了改进幅度。侧链RMSF:AF2仅0.15Å,AFsample2T提升至0.45Å,实验结构为0.58Å;骨架RMSF:AF2为0.10Å,AFsample2T为0.28Å,实验结构为0.30Å——AFsample2T的骨架RMSF中位值已非常接近实验结构,说明它采样到的是口袋骨架的物理运动范围,而不只是侧链旋转体的变化。均值口袋体积:AF2为209ų,AFsample2T为218ų,实验结构为256ų;但top 1%口袋体积方面,AFsample2T能达到389ų,远高于AF2的272ų。
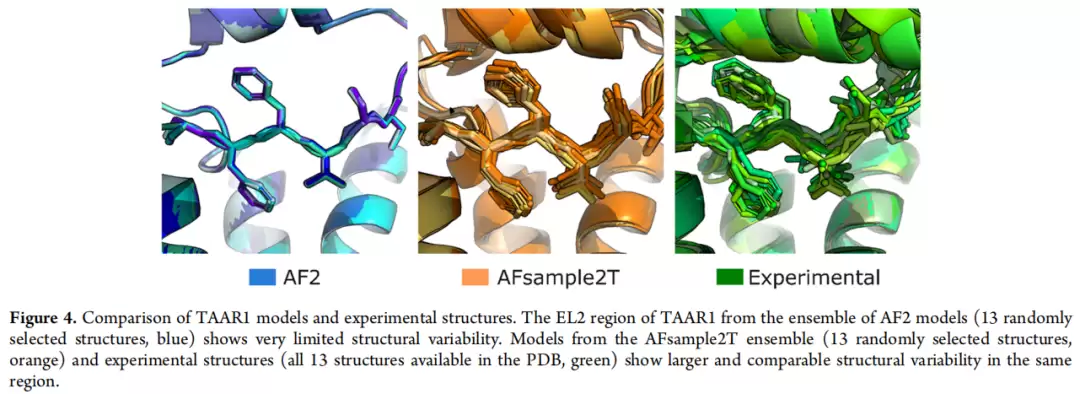
以TAAR1为例,EL2构象异质性的对比更为直观:AF2生成的所有13个模型EL2构象几乎完全重叠,而AFsample2T的13个随机抽样模型则展现出与13个实验结构相当的构象多样性。这一趋势在其余9个GPCR中均得到验证。μ-阿片受体的情况更为典型——AF2生成的口袋偏塌陷,AFsample2T集成中的口袋体积分布与实验结构更为一致,这也是AFsample2T在RMSD阈值1.15-1.30Å区间表现突出跳升的原因。
虚拟筛选:配体引导选模型可媲美实验结构
先看中位性能——不使用配体引导、随机选模型的情况下:实验结构的aLogAUC中位值为11.2,EF1%中位值为4.1;而AF2和AFsample2T的aLogAUC中位值分别仅4.4和4.2,EF1%中位值均为1.6。这个结果其实不令人意外,它与已有评估(Díaz-Rovira et al. 2023, Zhang et al. 2023)一致——没有配体引导时,实验结构的优势仍然显著。
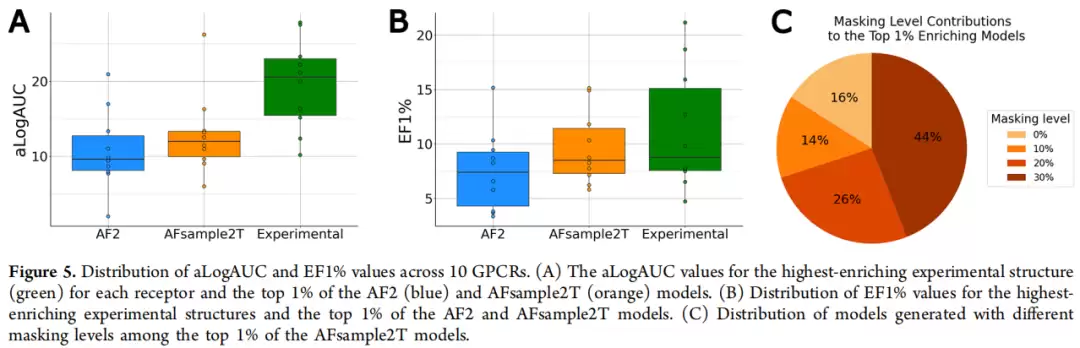
但转到top 1%性能(配体引导选模型)后,情况发生了质的变化。实验结构最优的aLogAUC为19.6,AF2 top 1%为10.8,而AFsample2T top 1%达到了12.9;EF1%方面,实验结构最优为11.3,AF2为7.5,AFsample2T为9.6。更具体的受体层面来看,TAAR1受体上AFsample2T top 1%的aLogAUC达到了32.0,甚至超过了实验结构最优的27.8;μ-阿片受体上AFsample2T为16.2,实验最优为15.2。在七个受体中(5-HT₁A、D₁、D₂、H₁、M₄、MT₁、TAAR1),AFsample2T的中位富集度也优于AF2。
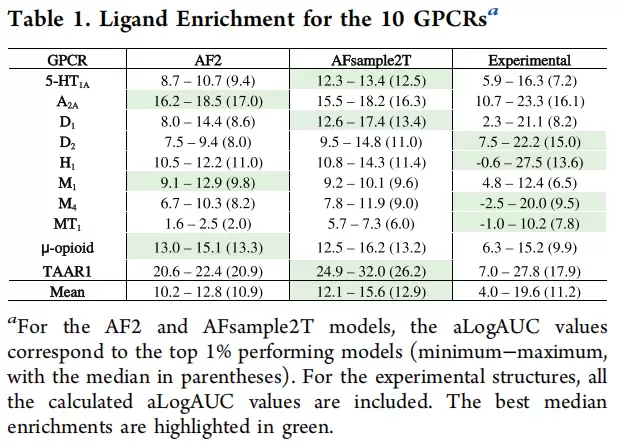
那么,什么样的蒙版概率最有效?分析top 1%模型的来源发现,20%蒙版贡献了44%,10%蒙版贡献26%,30%蒙版贡献16%,0%蒙版仅贡献14%。不同蒙版概率的互补贡献恰恰验证了集成策略的合理性——20%蒙版最多,但其他档位同样不可或缺。
模型数量分析
在计算成本方面有一个重要发现:分析10、100、250、500、1,000个模型的结果表明,生成100-250个模型即可获得接近1,000个模型水平的最大富集度。这对于实际应用的计算资源分配有直接的指导意义。
讨论:方法定位与适用边界
与相关方法的比较
| 方法 | 原理 | 优势 | 局限 |
|---|---|---|---|
| AFsample2T(本文) | 局部MSA列蒙版 | 构象多样性与模型置信度平衡;靶向效率高 | 依赖构象选择模型;不适用于强诱导契合 |
| 全局MSA蒙版(AFsample2) | 全MSA蒙版 | 适合捕获大构象变化(激活/非激活态) | 破坏整体折叠精度,口袋精度反而更低 |
| MSA浅采样(SPEACH_AF等) | 减少MSA行数 | 适合探索替代构象状态 | 同样侧重大范围构象变化 |
| MD精修 | 物理力场模拟 | 基于物理原理,可采样连续轨迹 | 膜蛋白系统计算代价极高;预测结构起点质量影响大 |
| AlphaFold3 / Boltz-2 | 配体-受体共折叠 | 直接预测复合物构象 | 结合位点建模精度尚待系统评估;依赖训练数据中相似配体 |
| 诱导契合对接(IFD) | 对接中允许受体灵活 | 针对特定配体的口袋适应 | 依赖单配体;不适用于scaffold-agnostic虚筛 |
方法局限性
话说回来,AFsample2T也有它的适用边界。首先,诱导契合场景受限——该方法基于构象预选模型,如果某个配体结合引发显著的诱导契合效应,预生成的集成可能无法覆盖相应构象。其次,AF2训练数据存在偏差,10个基准受体中有4个结构包含在AF2训练集中,模型对未知结构受体的泛化性能可能偏低。第三,无配体场景表现有限——中位富集度仍显著低于实验结构,对于完全无配体信息的孤儿受体,虚筛结果可靠性存疑。第四,受体构象能量估计未纳入,集成对接目前没有对不同构象进行能量权重校正。最后,这是GPCR特化设计,蒙版区域基于GPCR的保守拓扑定义,应用至其他靶标类别需要重新定义靶向区域。
方法延伸与展望
展望方面,有几个方向值得关注:将靶向蒙版指向GPCR变构口袋,有望生成适用于变构虚筛的构象集成;作者明确指出方法可迁移至激酶等具有保守折叠的靶标家族,源代码已开源;未来可将AFsample2T与AF2中的模板偏向策略结合,进一步提升特定功能构象的建模精度;随着AF3和Boltz-2等配体-受体共折叠方法的成熟,与AFsample2T的系统性对比将是重要研究方向。
实操指南:如何将AFsample2T用于GPCR虚筛
推荐工作流程(四步)
对于实际应用者,这里提供一个四步工作流程:第一步,生成AFsample2T集成——确定目标受体的激活/非激活态,定义靶向蒙版区域(胞外TM加EL2,参照GPCRdb编号),四档蒙版概率(0/10/20/30%)各生成至少62个模型,推荐集成规模不低于250个(计算资源充足时建议1,000个)。第二步,对接已知配体与诱饵分子——活性化合物至少10个已知配体(聚类后),诱饵分子每活性分子匹配至少50个(ZINC来源),对接工具推荐DOCK3.8或同类物理评分工具。第三步,配体引导模型筛选——计算每个模型的aLogAUC和EF1%,选取top 1%高富集模型(约10个模型/1,000集成),人工检查验证关键相互作用是否合理。第四步,使用1-3个top模型对目标化合物库进行全面对接。
决策树:实验结构 vs. AFsample2T
简单来说:如果存在受体实验结构,优先看是否有多个配体-受体复合物结构——有的话优先使用实验结构(通常一个结构即可达到top AFsample2T水平);如果只有一个或无配体结构,要看该结构的富集度是否良好,良好则使用实验结构,否则考虑用AFsample2T补充。如果没有任何实验结构,则使用AFsample2T加配体引导筛选——但注意,如果连已知配体都没有,那就是高风险场景,结果可靠性存疑。
回到问题的本质。这篇工作的核心贡献在于识别并缓解了AF2用于药物发现时的具体瓶颈——不是笼统地"增加采样多样性",而是有针对性地在结合口袋区域释放进化约束,并通过严格的回顾性虚拟筛选评估验证了方法的实用性。方法的优雅之处在于其极简的设计逻辑:仅需修改MSA输入(局部蒙版),无需改变AF2网络架构,无需额外训练,即可在结构质量与构象多样性之间实现有效的工程化权衡。
对药物发现社区的实践意义可以概括为四点:为无实验结构或实验结构质量有限的GPCR提供了可行的虚筛策略;证明了"集成加配体引导筛选"的范式可将AF2模型的虚筛性能提升至接近实验结构水平;提供了可直接复用的开源工具和方法学指南;方法可扩展至其他靶标类别,具有广泛适用性。
当然,局限性也需要时刻谨记:方法效果高度依赖配体引导的模型筛选——在无已知配体的场景中,AFsample2T的优势尚未被充分验证,仍需谨慎解读中位富集度数据。此外,方法与新兴共折叠工具(AF3、Boltz-2)的系统比较将是判断未来技术路线选择的关键。




