DRUGONE
在单细胞转录组研究中,一个长期存在的困惑是:基因表达量高,就一定意味着它功能重要吗?答案是否定的。许多关键转录因子表达量其实很低,却掌控着细胞的命运;而像核糖体或线粒体这类基因,虽然表达量极高,但往往与细胞身份的界定关系不大。这促使研究人员开发了一套名为SIGnature(Scoring the Importance of Genes)的新框架。其核心思路是:通过解读单细胞基础模型(single-cell foundation model)内部的attribution信号,来量化一个基因在特定细胞中的“功能重要性”。
SIGnature借助可解释人工智能(XAI)中的attribution方法,将每个基因对单细胞基础模型潜在表征(latent embedding)的贡献度进行量化,最终得到一个更具鲁棒性、且能在不同数据集间相互比较的基因重要性分数。研究团队进一步开发了相应的SIGnature软件包,使其能够在大规模单细胞图谱中实现快速的signature查询。
作为应用示范,研究人员利用该框架深入分析了严重COVID-19与脓毒症中发现的MS1单核细胞程序。通过对超过400项单细胞研究的搜索,他们发现这一程序与川崎病(KD)、噬血细胞性淋巴组织细胞增多症(HLH)以及发热伴血小板减少综合征(SFTS)等多种高炎症疾病存在关联。进一步的实验验证表明,KD患者的血清确实能够诱导MS1表型的形成。这项研究结果暗示,SIGnature不仅能提升单细胞signature评分的鲁棒性,更有望揭示不同疾病背后共享的免疫机制。

长期以来,研究者习惯于通过基因的表达量来判断其重要性。但绝对表达量这条“老路”常常会把人带偏。转录因子就是个典型例子,它们往往表达量很低,却是细胞命运的决定者;反过来,像MALAT1、核糖体蛋白和线粒体基因这类,则经常因为技术原因被检测出高表达,却不一定有多大的功能权重。
因此,目前单细胞分析常用的方法是依赖差异表达分析、GSEA、GSVA、Seurat或Scanpy等手段,通过相对表达的变化来推断功能。但这些方法普遍存在一个核心短板:不同实验间的批次效应、测序深度差异和细胞组成差异非常强烈,导致跨研究比较变得异常棘手。
与此同时,单细胞基础模型正在快速发展。这类模型通过海量数据预训练,能够学习到有生物学意义的潜在表征。但问题也随之而来——这些模型就像一个“黑箱”,研究者很难搞懂模型究竟认为哪些基因最重要。
那么,是否可以借助可解释AI中的attribution方法,把基础模型的黑箱表征,拆解成“基因功能重要性”的清晰答案?如果模型真的学到了生物规律,那么attribution得分高的基因,自然就应该对应那些决定细胞身份与功能的关键调控基因。
方法
为此,研究团队构建了SIGnature框架,它基于基础模型的attribution来计算单细胞层面的基因重要性。具体操作上,他们首先选定了多个主流单细胞基础模型,包括scFoundation、scGPT、SCimilarity、scTab和scVI等。模型需要满足两个基本条件:输入固定的基因集合,并能够输出具有生物学意义的细胞表征向量(embedding)。
接着,他们引入了可解释AI中几种常见的attribution方法,包括Integrated Gradients(IG)、Input × Gradient(IxG)和DeepLIFT(DL),用来计算每个基因对潜在表征的贡献值。为了让计算适配多维的表征向量,他们在模型末端添加了一个summation layer,使得embedding可以被映射成attribution向量。
研究团队在多个单细胞数据集上进行了系统性的基准测试,从attribution的运行速度、抗技术噪声的能力、标志基因的富集效果,以及跨研究的鲁棒性等维度进行了全面评估。最终,他们选定了“SCimilarity + Integrated Gradients”作为后续分析的主力组合。
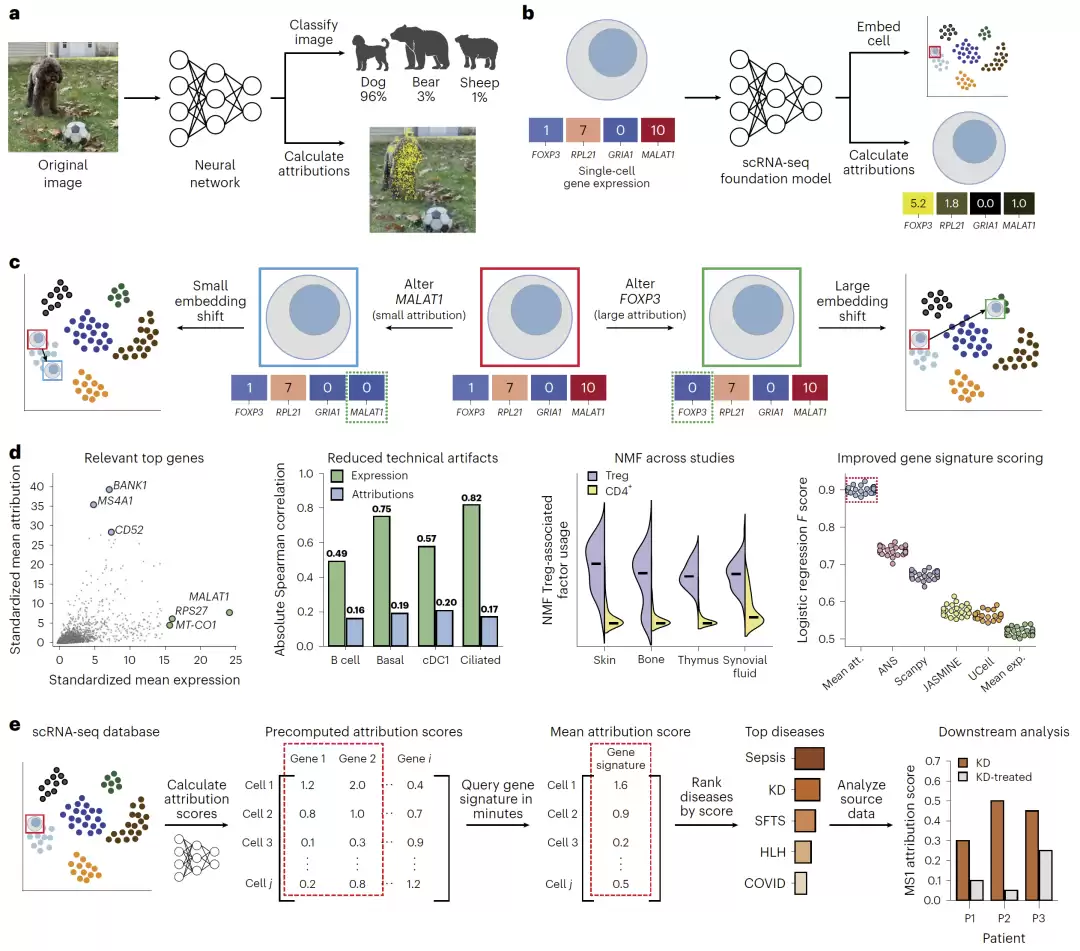
图1:SIGnature框架概念图与attribution在单细胞基础模型中的作用机制。
结果
Attribution 能够量化单细胞中的“基因重要性”
首先,研究团队验证了attribution是否真的能反映生物学功能。结果相当清晰:在B细胞中,attribution得分最高的基因包括BANK1、CD79A和MS4A1,这些都是经典的B细胞标志基因。而相比之下,表达量最高的基因则主要是MALAT1、MT-CO1和RPS27这类技术相关的高表达基因。
这一趋势在多种肺细胞类型中同样成立。研究显示,相比于基于表达量的排序,attribution排序能够显著提升标志基因和转录因子的排名。
更进一步,对CD4+ T细胞的分析揭示了attribution的“精准度”:
- GATA3在Th2细胞中attribution更高;
- RORC在Th17细胞中attribution更高;
- FOXP3在Treg细胞中attribution更高。
结论很明确:attribution更像是在衡量“功能调控重要性”,而不仅仅是表达量的高低。
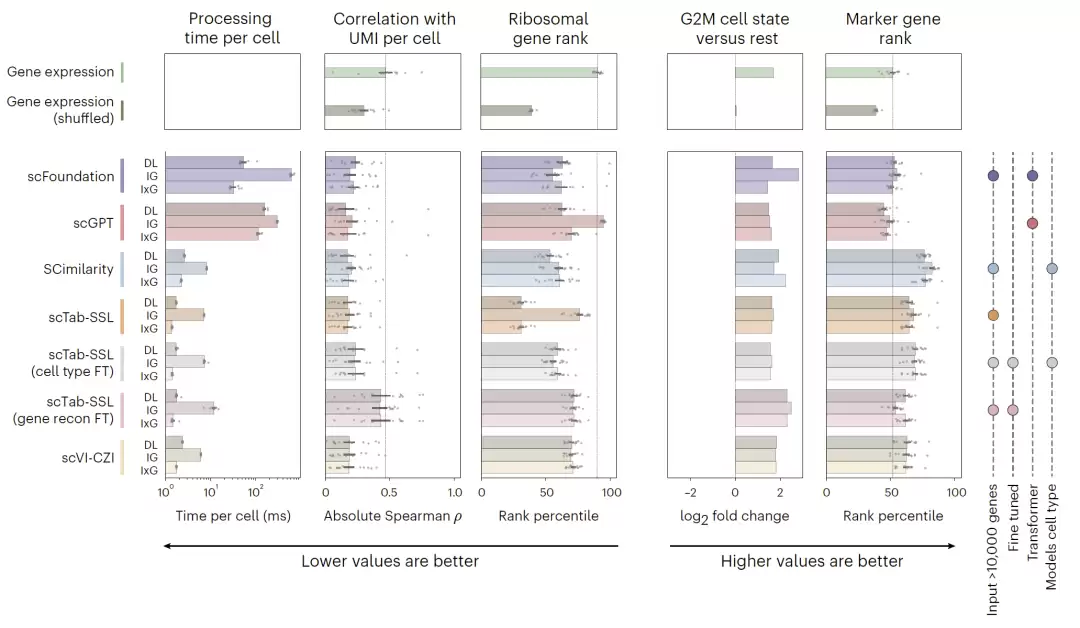
图2:Attribution与传统表达量在marker gene和转录因子识别中的对比。
Attribution 显著降低技术噪声影响
接下来,研究团队考察了attribution对技术伪影(artifact)的抗性。结果显示,与表达量相比,标志基因的attribution与UMI计数、检测到的基因数等测序深度指标的相关性明显更低。
举一个具体的例子:在非经典单核细胞(nonclassical monocyte)中,
- 表达量(expression)与测序深度的相关性高达ρ=0.71;
- 而attribution的相关性仅为ρ=-0.12。
为了进一步验证,他们模拟了数据缺失(dropout)的情况,随机移除了50%的counts。结果显示,attribution排名前列的基因依然保持了93%的重叠率,说明它对数据缺失具有很强的鲁棒性。
此外,针对不同基础模型的基准测试也表明,attribution在所有模型中都能有效降低核糖体基因的重要性,同时增强有丝分裂相关基因和细胞类型标志基因的信号。
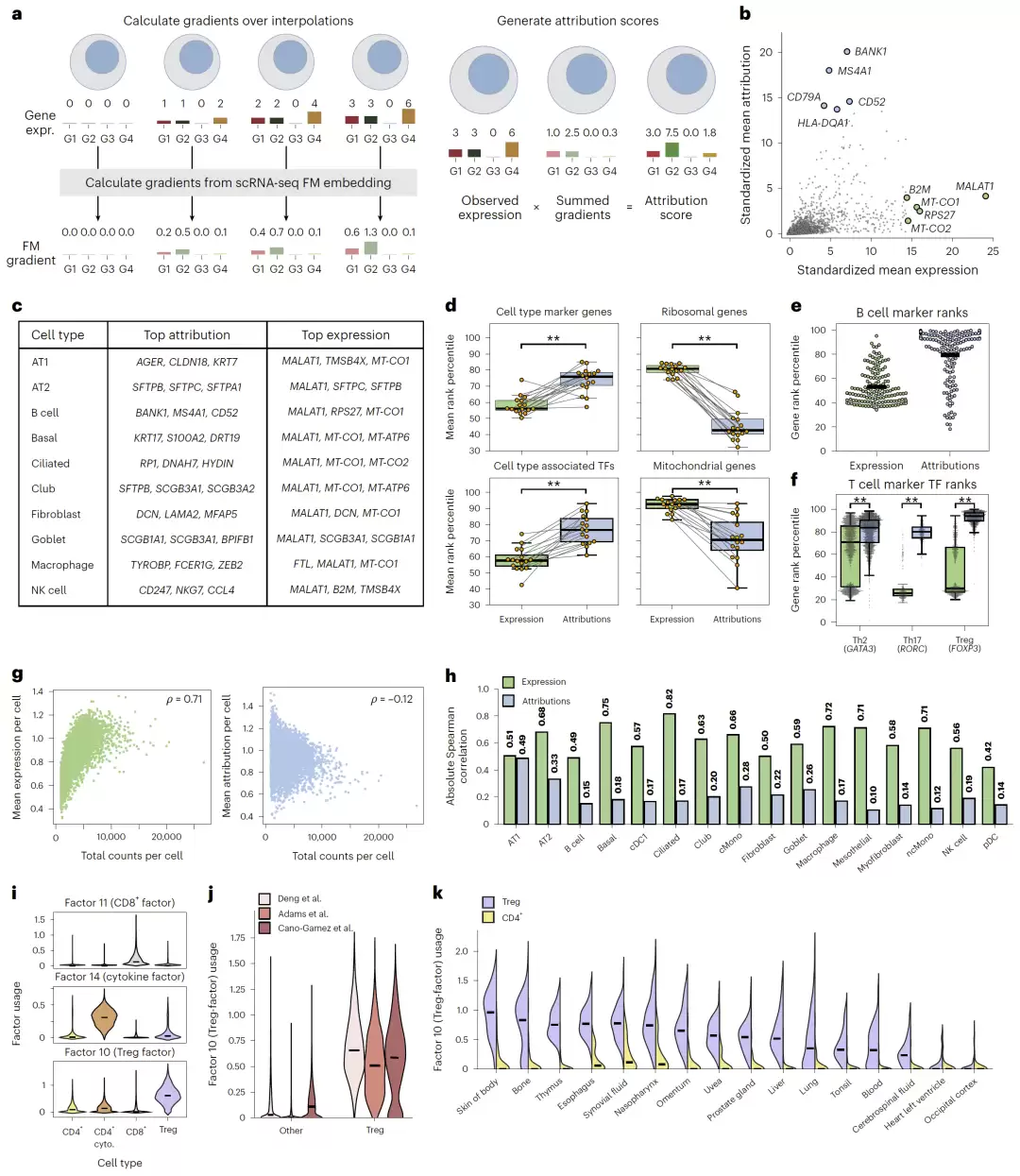
图3:不同foundation model attribution的benchmark与抗技术噪声能力分析。
Attribution 支持跨研究基因程序发现
研究团队随后测试了attribution是否能够支持跨数据集的基因程序发现。他们将多个T细胞数据集拼接后,对attribution矩阵进行了非负矩阵分解(NMF)。
结果令人振奋:他们成功恢复了多个具有明确生物学意义的基因程序,包括:
- CD8+ T细胞因子;
- 细胞因子反应因子;
- Treg相关因子。
其中,Treg因子富集了FOXP3与IL2RA等经典的调控基因,并且能够推广到来自16种不同组织的Treg细胞。与基于表达量的NMF相比,基于attribution的NMF更稳定、受研究特异性效应的影响更小,也更容易恢复有生物学意义的因子。
值得关注的是,基于attribution的方法甚至能达到接近有监督的scETM模型的效果,而且不需要重新训练。
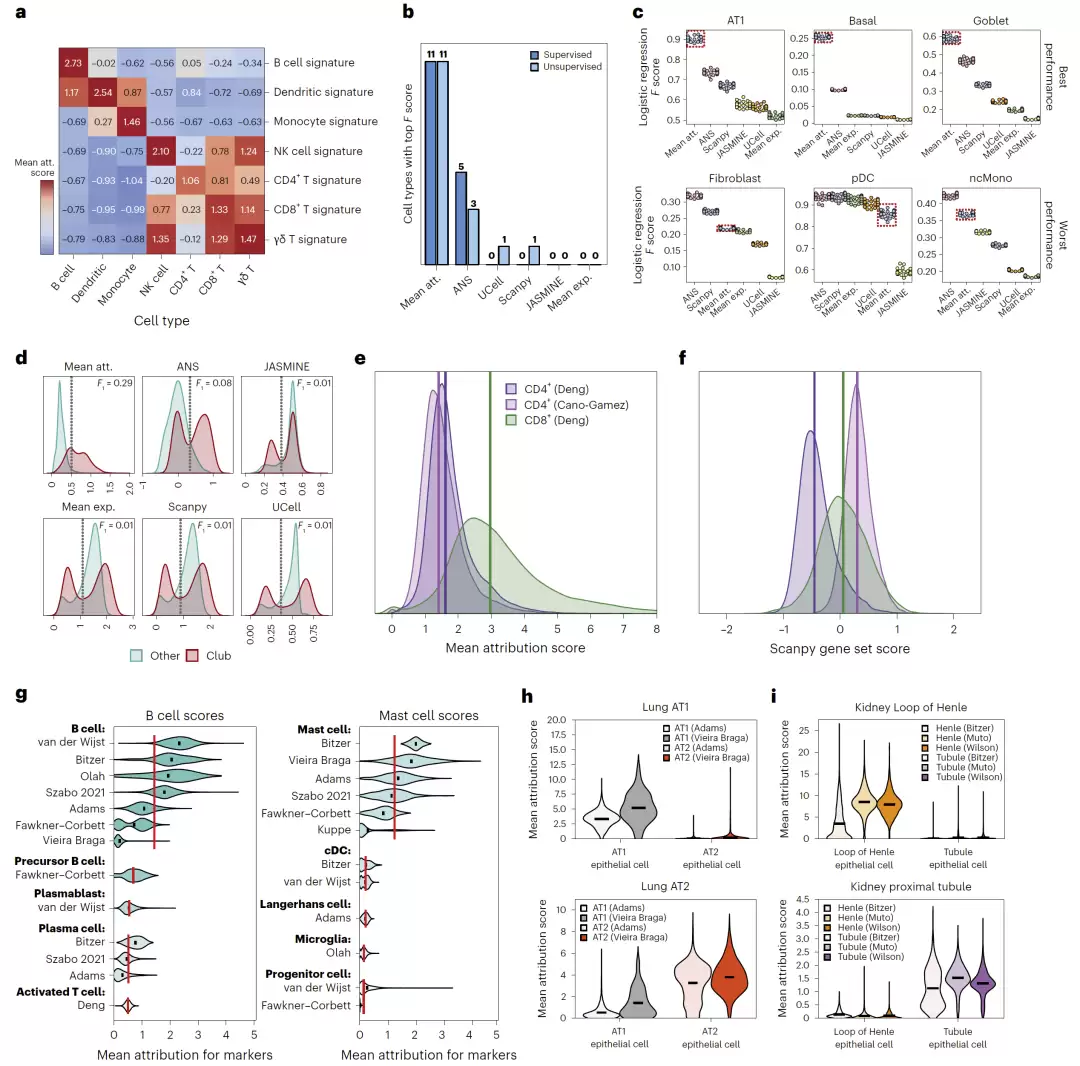
图4:基于attribution的跨研究NMF基因程序分析。
Attribution 显著提升 signature 评分能力
接下来,研究团队将attribution应用于基因signature的评分。他们发现,对一个signature中所有基因的attribution求平均值,可以有效地衡量该signature在单细胞中的激活程度。
在PBMC数据集中的测试显示:
- B细胞signature在B细胞中得分最高;
- NK细胞signature在NK细胞中最高;
- CD8+ T细胞signature在CD8+ T细胞中最高。
进一步与Scanpy、UCell、JASMINE和ANS等方法比较,基于attribution的评分方法(mean attribution)在32项测试中赢得了23项,在有监督和无监督任务中均表现最佳。
更关键的是,基于attribution的评分在跨研究分析中表现出极强的鲁棒性。例如,传统方法Scanpy可能会错误地给某些CD4+ T细胞打出比真实CD8+ T细胞还高的CD8 signature得分,而attribution方法则不会出现这个问题。研究团队在120万个细胞、15个独立实验中对这一结论进行了验证。
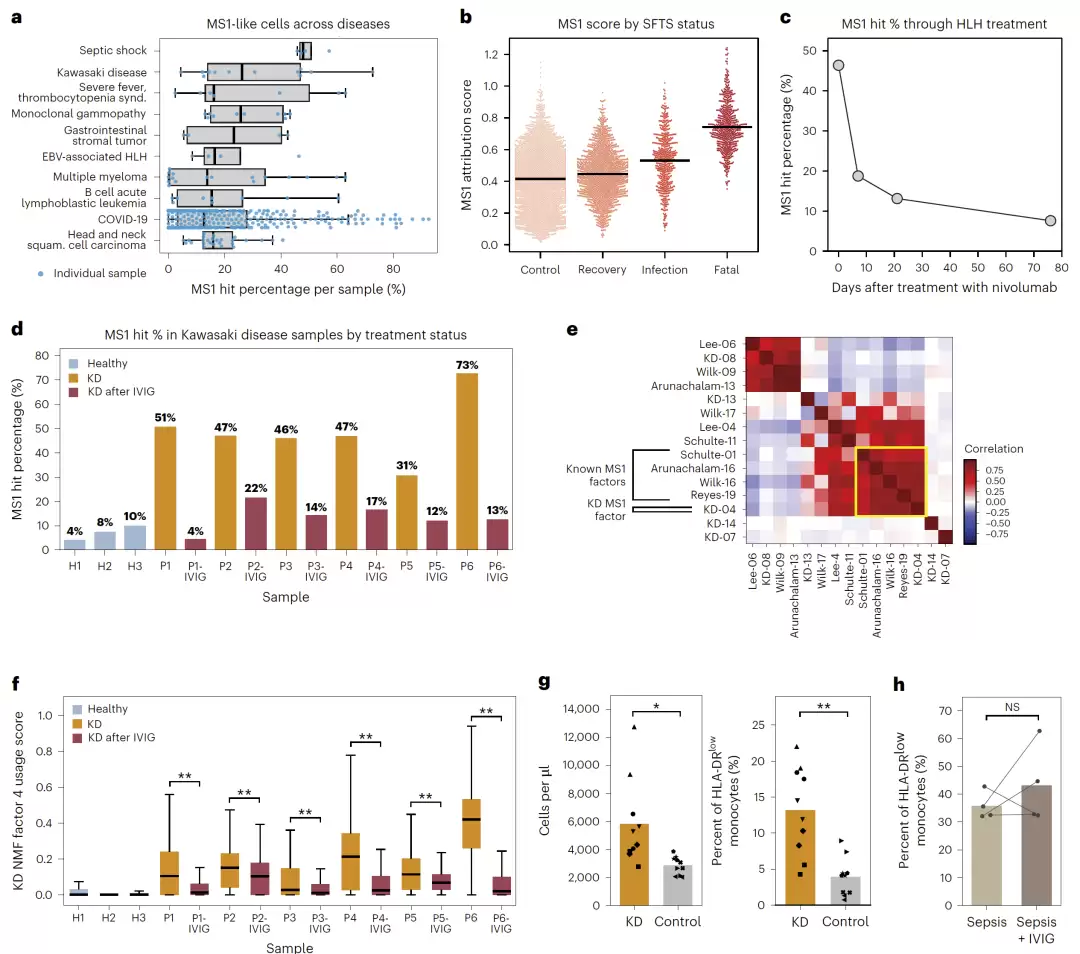
图5:Attribution-based signature scoring与传统方法的系统比较。
SIGnature 在2200万细胞中发现共享炎症状态
最后,研究团队展示了SIGnature在大规模应用场景下的真正实力。由于attribution可以预先计算,他们得以在2200万个细胞中快速搜索特定的基因signature。
他们选择了严重COVID-19与脓毒症中的MS1单核细胞signature,并在412个疾病研究中进行了搜索。结果不仅重新发现了此前已知的脓毒性休克和严重COVID-19,还意外地发现它与川崎病、发热伴血小板减少综合征(SFTS)以及噬血细胞性淋巴组织细胞增多症(HLH)也存在关联。
在川崎病的数据中,MS1样细胞在接受IVIG治疗后显著减少。进一步的实验验证表明,川崎病患者的血清能够诱导紧急髓系生成(emergency myelopoiesis),并增加HLA-DRlow的MS1样单核细胞比例。这一结果说明,SIGnature不仅能完成signature评分,还能发现此前未知的疾病关联机制。
讨论
总的来说,SIGnature框架首次将可解释AI的attribution系统性地引入单细胞基础模型的解释中,构建了一套统一且可扩展的“基因重要性评分体系”。相比于传统的基于表达量的方法,attribution更关注那些真正驱动模型潜在表征的关键基因,因此能够增强转录因子和标志基因的信号,同时有效降低测序深度与技术伪影的干扰。研究结果表明,基于attribution的表征不仅能改善跨研究的基因程序发现,还能在超大规模图谱中实现分钟级的signature查询,从而揭示出不同疾病之间共享的免疫状态。可以预见,这类“可解释的基础模型”未来将成为单细胞分析领域的重要基础设施,让AI不再只是一个预测工具,而是真正成为帮助研究人员理解细胞调控机制的得力助手。
参考资料
Gold, M.P., Reyes, M., Diamant, N. et al. Scoring gene importance by interpreting single-cell foundation models. Nat Biotechnol (2026).
https://doi.org/10.1038/s41587-026-03112-5




