
先分享一个我们最近读到的判断,它可能颠覆很多人的直觉。近年来,机器学习的热潮确实席卷了小分子药物发现领域,各种新模型、新算法层出不穷,让人感觉“新药研发的黄金时代”近在眼前。但牛津大学 Charlotte Deane 课题组的一篇 Perspective,却以罕见的冷静与犀利,为这股热潮泼了一盆冷水。
作者给出的诊断非常直接:在小分子药物发现这个领域,性能提升的瓶颈并非算法本身的精巧程度,而是喂养算法的数据——其质量和数量,远远没有跟上。这个判断,对于那些热衷于追逐模型新颖性的研究范式而言,无疑是一次及时且必要的反思。
一、背景:一边是巨大挑战,一边是满载期望
1.1 小分子药物的核心地位与开发痛点
大家可能有一个直观感受:市面上绝大多数药片、胶囊,核心成分都是小分子。数据也证实了这一点:在全球已获批的药物中,小分子化合物占比高达 90%,是现代医学的绝对支柱。它们分子量小,能轻松穿透细胞膜,直击蛋白质或 RNA 靶点。不仅如此,近年的应用还在向 PROTACs、分子胶水等新方向拓展。
但挑战同样巨大。有一个被称为“Eroom 定律”的趋势,听起来有些令人沮丧——从 1950 年到 2012 年,每花 10 亿美元能获批的新药数量,每 9 年就减少一半。这几乎是 Moore 定律的“反向版”。一款新药从实验室走到药房,动辄数年甚至十年,烧掉的钱以亿计。背后的痛苦,每一位从业者都深有体会。
1.2 机器学习为何被寄予厚望?
因为机器学习在邻近几个领域表现得太出色了,让人很难不把希望投射过来:
- 计算机视觉中的扩散模型、对比学习,催生了 DALL-E、Stable Diffusion;
- 自然语言处理中的 GPT-4、LLaMA 2,能力已接近人类;
- 蛋白质结构预测的 AlphaFold 2,在 CASP14 竞赛中比第二名精度高出 2.6 倍,那是现象级的突破。
所有这些成功的背后,都有一个共同点:数据足够丰富,再加上精巧的架构,两者缺一不可。这自然让人们期待,同样的奇迹也能在小分子领域上演。
事实上,机器学习已经渗透到小分子药物发现的多个环节:
| 任务 | 描述 |
|---|---|
| 从头分子设计(De novo design) | 生成具有目标性质的新颖分子 |
| 逆合成预测(Retrosynthesis) | 规划高效的合成路线 |
| 分子对接(Docking) | 预测配体与靶点结合的三维构象 |
| 性质预测(Property prediction) | 估计 ADMET、活性等关键分子性质 |
二、核心发现:算法在“内卷”,性能却未明显提升
2.1 基准测试揭示的残酷现实:进步微乎其微
为了弄清真实进展,作者翻出了三个经典基准测试,将历年发表的方法性能指标与发表时间做了相关性分析。结果令人扎心:
| 基准测试 | 任务 | 指标 | 时间-性能相关性(ρ) | 结论 |
|---|---|---|---|---|
| CASF-2016 | 蛋白质-配体结合亲和力预测 | Pearson"s R | 0.03 | 近乎零相关,无进步 |
| HIV MoleculeNet | HIV 蛋白酶活性分类(QSAR) | AUROC | 0.09 | 近乎零相关,无进步 |
| USPTO-50k | 单步逆合成预测 | Top-1 准确率 | 0.87 | 有增长,但仍是渐进式的 |
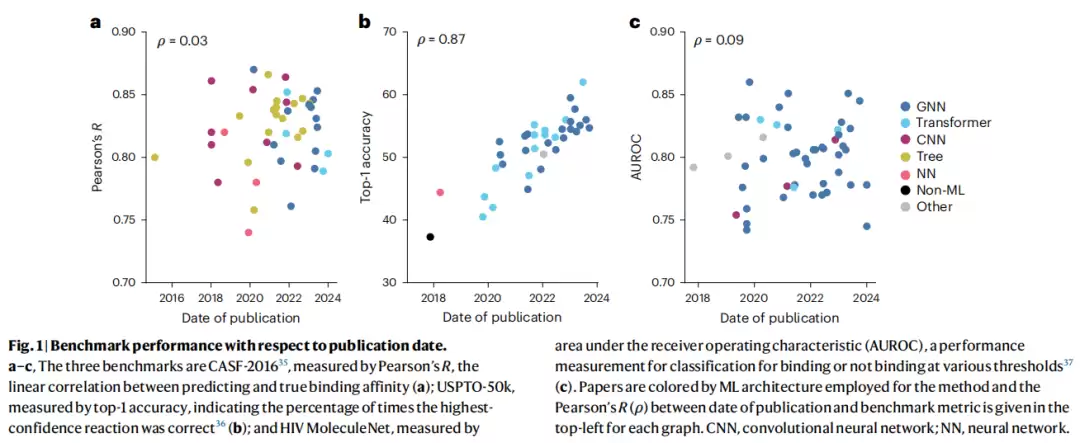
注意,即便是表现最好的 USPTO-50k,也只是渐进式提升,远未达到 AlphaFold 2 那种数量级的跃升。换句话说,算法在“内卷”,但成绩单变化不大。
2.2 先进架构为何水土不服?
具体到几种被寄予厚望的架构,情况同样不容乐观:
图神经网络(GNN) 理论上最适合小分子,能直接编码分子图结构。它在材料发现(GNoME)甚至新型抗生素发现上都有成功案例。但在小分子药物发现的全面评测中,它与传统方法相比,并未展现系统性优势。
等变图神经网络(Equivariant GNN) 将三维对称性内嵌进去,省去了数据增强的麻烦。然而研究表明,这一架构进步同样未带来精度或泛化性的显著提升,其价值更多体现在提供了更可解释的预测归因。
扩散模型(Diffusion models) 在图像生成领域大放异彩。当它被应用于分子对接(DiffDock)和分子生成时,精度确实比旧方法高了一截。但很快,PoseBusters 和 PoseCheck 两项独立研究就指出了问题:这些模型生成的分子构象在物理化学层面并不合理,与蛋白质的相互作用也存在明显缺陷。
作者态度明确:未来那些看起来很酷的新方法(比如一致性模型、流匹配),如果只是“开箱即用”地套用,同样很难带来质的飞跃。真正的进步,需要架构创新、工程决策和高质量数据三者的协同,缺一不可。
三、数据量问题:一道根本性的规模鸿沟
3.1 小分子数据的先天不足
机器学习的本质是从数据中寻找规律,数据量越大,模型能学到的东西就越强。这个“定理”到了小分子领域,却撞上了现实的墙:
- 实验数据的生成成本高昂、劳动密集,不可能像互联网数据那样被动地大规模积累。
- 制药公司将大量数据视为核心知识产权,严密封锁,形成了一座座数据孤岛。
- 公开数据集的规模仅有数万到数十万量级,与隔壁领域完全不在一个量级。
对比一下这些数字,就能看出差距有多大:
小分子 ML 数据集: 数万 ~ 数十万 蛋白质序列数据库(UniRef):数亿 图像生成训练数据(DALL-E 2):数亿 大语言模型训练数据(LLaMA 2):数万亿 token
3.2 结构数据的特殊困境
对接算法、结构生成模型、打分函数都需要蛋白质-配体复合物的三维实验结构。PDBBind 是最重要的公开数据集,但即使是 2020 版本,也仅收录了 19,443 个复合物——这个规模直接设定了相关模型性能的天花板。
现有的一些扩充策略,比如交叉对接增强、合成配体生成、自蒸馏等,虽有一定帮助,但短期内想大幅增加结构数据量或多样性,难度很大。
3.3 负面数据:严重缺失的信息“暗面”
在小分子研究中,负面数据(比如无活性的化合物、失败的化学反应)几乎不公开。原因很现实:
- 发表偏倚:期刊更喜欢阳性结果。
- 知识产权保护:失败的实验数据也可能泄露研究方向。
- 整理成本高:负面数据往往分散且不规范,整理起来费时费力。
这直接导致训练数据中正负样本严重失衡。比如在产率预测任务里,模型根本学不到失败反应的规律,预测的可靠性大打折扣。潜在解决路径包括改革学术发表规范、生成合成负样本,以及从博士论文、实验室记录中挖掘“暗数据”。
3.4 希望在哪里?新兴的数据来源
众包药物发现 是一个很好的方向。COVID Moonshot 项目堪称标杆案例,针对新冠病毒 Mpro 蛋白酶,以完全开放的方式协作,最终产出了 470 个晶体结构、两千多个化合物的 IC₅₀ 数据、三千多个化合物的合成数据——全部公开。
联邦学习(Federated Learning) 则提供了另一种思路:在数据不转移的前提下共享模型知识。MELLODDY 项目和 Effiris 项目的结果都很积极,模型适用域分别扩展了 10% 和 83%。
四、数据质量问题:噪声、偏差与假阳性
4.1 数据偏差:模型学到了“幻象”
机器学习模型的忠实度有些过头:它会一丝不苟地学习训练数据里的所有规律——包括那些并不反映真实物理化学规律的“幻象”。
- 人为偏差:数据并非随机采样,而是由研究者主动选择,往往偏向熟悉的化学空间和合成路线。
- 归纳偏差:最典型的是基于 ML 的虚拟筛选工具,研究发现它们只依赖配体层面的特征,本质上是在做“配体-配体相似性”比对,而非真正学习蛋白质-配体相互作用的规律。
研究者提出的“不对称验证嵌入”方法,通过更严苛的训练-验证集划分来惩罚这类模型,结果发现许多被广泛引用的方法,其真实准确率远低于论文中报告的数值。
4.2 数据噪声:标签的不确定性被严重低估
小分子数据集里的噪声,比大多数人想象的要严重得多。
实验条件不一致:最典型的是将来自不同实验室、不同测定条件的 IC₅₀ 数据混在一起,训练一个 QSAR 模型。但问题在于,不同测定条件让这些数据根本不可比。同一靶点、同一化合物,不同实验室的 IC₅₀ 值之间,R² 仅有 0.53;即便使用相对更一致的 Kᵢ 值,R² 也只有 0.81。研究估计,CASF-2016 上 ML 打分函数的理论精度上限只有 Pearson"s R 0.76——这个上限并非模型缺陷所致,而是数据噪声的固有极限。
假阳性:泛筛干扰化合物(PAINs)
更令人震惊的是,作者还指出:在广泛使用的 HIV MoleculeNet 数据集中,404 个“真实活性”化合物里,有 70% 命中了 PAINs 子结构过滤规则。这意味着该基准测试中相当比例的“真实信号”,可能只是实验噪声。另一个案例是,在常用的元学习基准中,仅凭一个化合物命中多少靶点(这是 PAINs 的典型特征),就能高度预测它的“活性”——这说明数据集本身已被噪声严重污染。
五、验证体系的问题:自我感觉良好的幻觉
5.1 现有验证范式的系统性缺陷
当前小分子 ML 方法的验证,通常遵循这样一个流程:提出新架构 → 在流行训练集上训练 → 在流行基准测试上评估 → 报告(通常微小的)提升。这一范式有一个根本问题:测试数据和训练数据高度同质,而真实的药物发现中,模型面对的是分布外(OOD)数据。
两个例子很有说服力:
案例一:分子对接的“重对接”问题
ML 对接方法往往以“重对接(Redocking)”作为评测标准:给定一个已知的晶体结构,预测配体构象。但问题是,如果晶体结构已知,为何还需要对接?当研究者在更真实的场景下评测(比如对接入预测蛋白质结构),模型性能会大幅下滑。重对接给出的是一个过于乐观的估计。
案例二:逆合成的单步与多步脱节
USPTO-50k 评测的是单步反应预测的准确率。但实际使用中,逆合成工具需要找到完整的多步合成路径。最新研究发现,单步方法在基准上的准确率,与它在完整路径搜索中的成功率之间存在明显脱节。
5.2 哪些改进方向值得关注?
作者展示了一个发人深省的例子:一个简单的基于决策树的打分函数,只使用配体层面的特征(即只能学习数据集偏差),在多个基准上与复杂的 GNN 方法性能相当。这说明,相当一部分“模型进步”来自于学习数据集偏差,而非真正理解物理化学规律。
因此,消融实验应更系统地推广,明确区分架构改进、数据改进和训练策略改进各自的贡献。更严格和真实的基准体系也在建设中:
| 机制 | 代表项目 | 特点 |
|---|---|---|
| 在线排行榜 | Therapeutic Data Commons (TDC) | 集中跟踪发表进展,提供细粒度任务分析 |
| 盲测竞赛 | Drug Design Data Resource (D3R) | 验证集保密,避免过拟合基准 |
| 盲测竞赛 | CACHE(命中筛选计算方法评估) | 公开结果,支持方法横向比较 |
六、与 AlphaFold 2 的对比:为何差距如此巨大?
AlphaFold 2 的成功并非靠某一个关键创新,而是多个精心设计的组件协同作用的结果:不变点注意力、迭代精化策略、利用序列数据补偿结构数据的不足……而在小分子领域,类似这种规模庞大、信息丰富的“杠杆数据”尚不存在。没有一个数据宝藏,可以让模型通过间接学习来弥补直接实验数据的稀缺。
这就是小分子 ML 与蛋白质结构预测之间最根本的区别。
七、总结与展望:在期望与现实之间,路在何方?
核心论断与建议
| 方面 | 现状 | 作者建议 |
|---|---|---|
| 算法 | 新架构不断涌现,但性能提升有限 | 与其追逐新架构,不如深入分析现有方法的瓶颈 |
| 数据量 | 公开数据规模远不足以支撑深度学习 | 推进联邦学习、众包、暗数据挖掘 |
| 数据质量 | 噪声高、偏差多、负样本缺失 | 建立更严格的数据清洗流程,纳入不确定性建模 |
| 验证体系 | 基准测试与实际应用脱节 | 推广消融测试、盲测竞赛、更真实的测试集 |
对研究者的几点启示
- 在发表新方法前,认真思考:性能提升究竟来自架构创新,还是更多或更好的数据?抑或只是学会了更多数据集偏差?
- 负面结果同样有价值。记录并发布失败实验,对推动领域共识至关重要。
- 计算研究者与实验研究者需要更紧密地合作。许多数据质量问题,如 PAINs、实验条件差异,都需要领域专知才能识别和处理。
- 将“是否在分布外数据上泛化”作为核心评测标准。在真实的药物发现场景中,模型几乎总是面对分布外数据。
写在最后
这篇 Perspective 的价值,不在于提出了什么革命性的新方法,而在于用扎实的数据分析揭示了一个常被忽略的事实:在小分子药物发现领域,我们对机器学习的期望,远远超前于支撑这些期望所需的数据基础设施。
算法的进步是必要的,但不是充分的。如果领域内的资源继续主要流向追逐更新颖的架构,而忽视数据生态系统的建设,那么“AlphaFold 时刻”在小分子药物发现中,或许还要等上很久。
数据驱动,才是正道。




