一、研究背景与问题
八元氮杂环结构在天然生物碱及人工合成分子中并不少见,它们常常发挥关键作用——蛋白激酶抑制、抗癌、麻醉受体相互作用、杀虫等生物活性涵盖广泛。这类稠合多环衍生物因其独特的三维刚性骨架,还被广泛用于配体、功能染料、荧光探针、半导体和光伏材料。前景虽好,但问题随之而来:中等环尺寸的氮杂环构建起来绝非易事。不利的熵效应与焓应变,使得这类骨架的合成极具挑战性。
另一方面,功能化喹啉是氮杂环家族中的“明星分子”。尤其是2-烯基取代喹啉,不仅可作为多功能合成中间体,还拥有抗疟、抗利什曼病、哮喘抑制及CysLT1拮抗等多种药理活性。尽管已有Wittig反应、2-甲基喹啉与醛/亚胺的缩合、喹啉N-氧化物直接乙烯化等方法,但开发更简洁、高效、绿色且通用的合成路线,依然是该领域的迫切需求。
在C−H键活化领域,自由氨基作为导向基团具有独特优势:它广泛存在于廉价原料中,配位能力强,更重要的是,氨基的显著亲核性可引发多种后偶联反应。然而,烯基C(sp2)−H键的直接官能化,相比于芳基C(sp2)−H键,仍是一块更难啃的骨头。
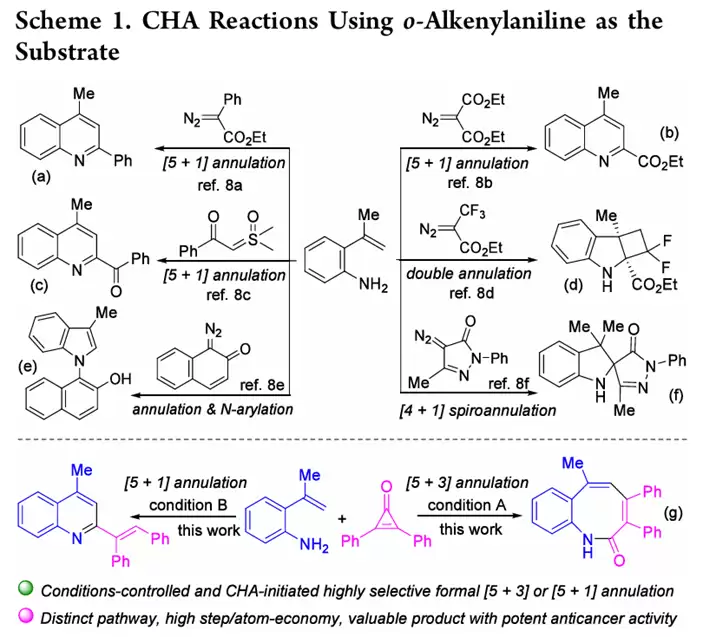
已有研究曾借助2-烯基苯胺底物,利用氨基作为自由基实现烯基C(sp2)−H键官能化。但已报道的工作多聚焦于重氮化合物或亚砜鎓叶立德作为偶联伴侣,主要生成六元或五元氮杂环骨架。因此,将偶联伴侣从重氮化合物拓展至环丙烯酮,并探索在不同条件下选择性合成不同类型杂环产物,成为本文要解决的核心问题。
二、文章亮点
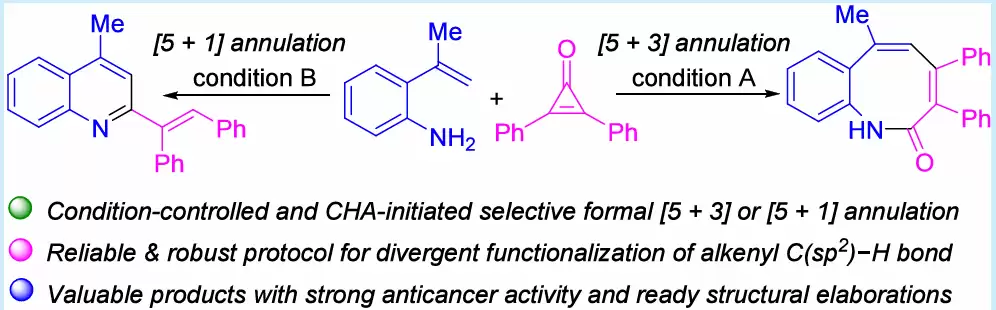
条件可控的选择性:本文巧妙通过改变反应条件——溶剂、添加剂、温度——从相同起始原料出发,精准切换合成路径,选择性获得苯并[b]氮杂辛酮(product 3,[5+3]环化)或2-烯基喹啉(product 4,[5+1]环化),实现了真正的多样性导向合成。
首创性偶联伴侣:本文首次将环丙烯酮引入2-烯基苯胺的Rh(III)催化C−H键活化体系,突破了对重氮化合物或亚砜鎓叶立德的依赖,且安全性更优。
卓越的原子经济性:反应放大至3 mmol规模,仍可获得中等至良好收率(3a:53%,0.64 g;4a:40%,0.46 g),实用性突出。
广泛的底物适用范围:苯并[b]氮杂辛酮(3a−3hh)和2-烯基喹啉(4a−4z)两类产物均展现出出色的底物普适性,多种供电子/吸电子取代基及不同位置取代的底物均可顺利兼容。
令人关注的抗癌活性:苯并[b]氮杂辛酮衍生物对SW-480、HCT-116、HeLa三种人癌细胞株表现出强抑制活性,与阳性对照5-FU相当,具备良好的先导化合物开发潜力。有趣的是,2-烯基喹啉产物4在此次测试中未显示显著抗增殖活性。
多样的产物修饰潜力:3a和4a可进一步转化为2-氯苯并[b]氮杂辛、3,4,6-三苯基苯并[b]氮杂辛、2-苯甲酰喹啉及多环产物等,结构拓展性极佳。
三、主要内容
反应条件优化
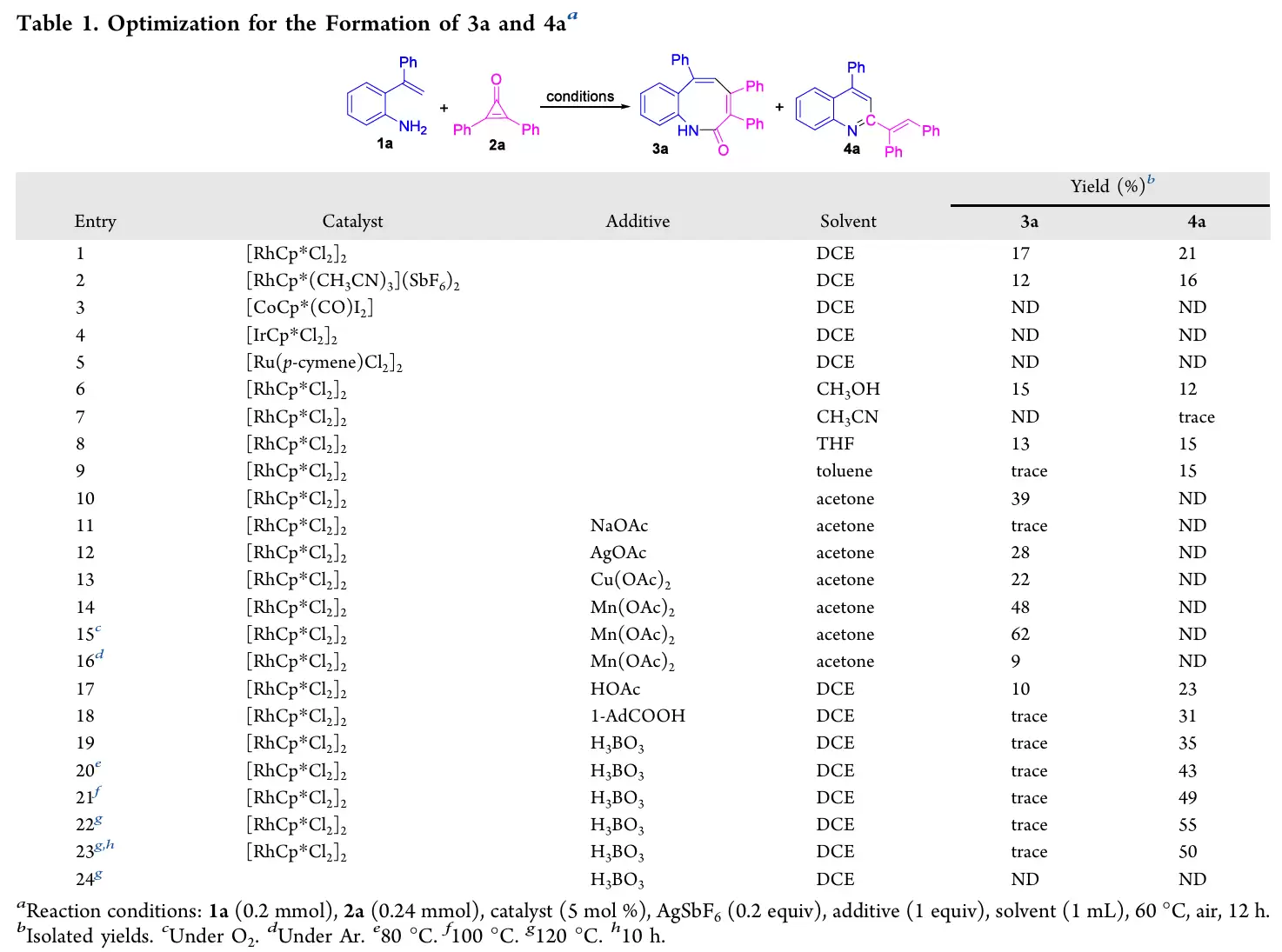
作者以1a与2a为模型底物,[RhCp*Cl2]2/AgSbF6为催化体系,系统筛选了催化剂、溶剂、添加剂及反应温度。关键发现可总结如下:
- 催化剂方面,[RhCp*Cl2]2效果最优,而[CoCp*(CO)I2]、[IrCp*Cl2]2、[Ru(p-cymene)Cl2]2基本无效。
- 合成3的最佳条件(条件A):丙酮为溶剂,加入1当量Mn(OAc)2,在氧气气氛下60°C反应12小时,3a收率达62%,且无4a生成。
- 合成4的最佳条件(条件B):DCE为溶剂,加入1当量H3BO3,在空气气氛下120°C反应12小时,4a收率为55%,3a仅痕量。
- 关键控制实验:无催化剂时反应完全不发生,证明Rh(III)催化不可或缺。
底物适用范围
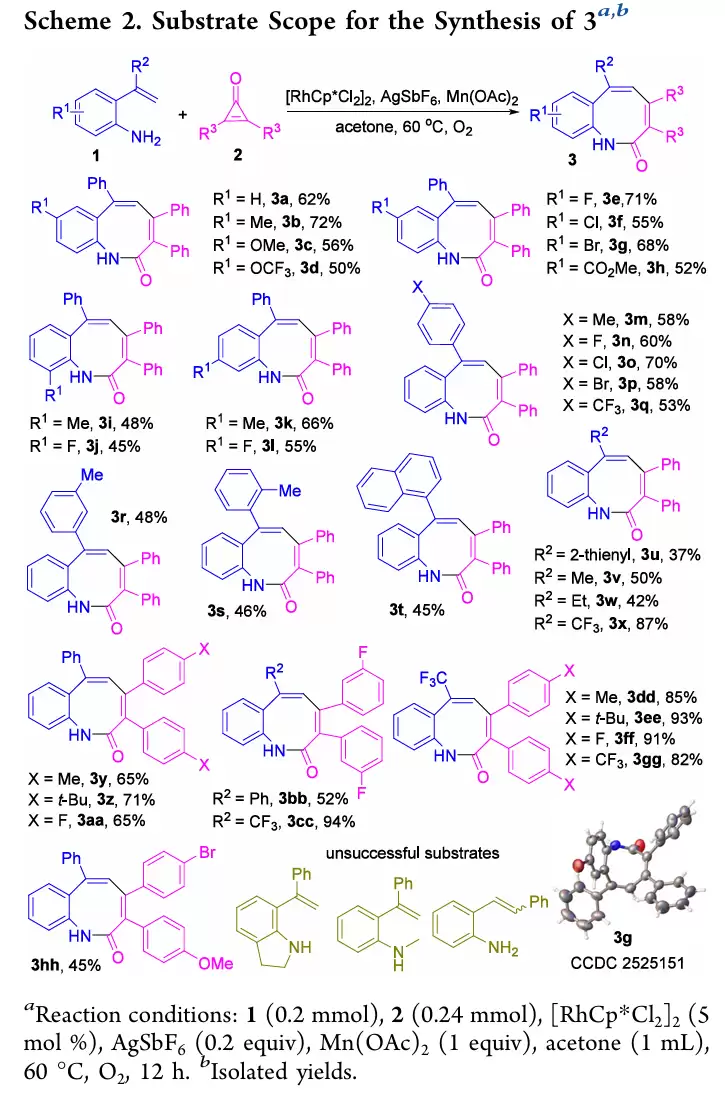
苯并[b]氮杂辛酮(product 3)系列:苯胺环对位无论是供电子基(Me、OMe)还是吸电子基(OCF3、F、Cl、Br、酯基),底物1均可顺利反应,以中等至优秀收率得到3a−3h。邻位(2-Me、2-F)、间位(Me、F)取代底物同样适用(3i−3l)。R2位可为取代苯基、萘基、2-噻吩基,甚至脂肪烷基(Me、Et、CF3)也均可兼容(3m−3x)。环丙烯酮2适用范围也很广,涵盖供电子基(Me、t-Bu)和吸电子基(F),以及不对称环丙烯酮(3hh)。值得注意的是,2-(3,3,3-三氟丙-1-烯-2-基)苯胺与环丙烯酮反应,收率高达82−94%(3cc−3gg)。
2-烯基喹啉(product 4)系列:苯胺环上对位、间位、邻位含Me、OMe、F、Cl、Br、CF3、NO2等取代基的底物,均能通过[5+1]环化顺利得到4a−4k。特殊底物如苯并二氧戊环胺、萘胺衍生物,以及R2为取代苯基或甲基的底物也均适用(4l−4t)。更有趣的是,2-(环戊-1-烯-1-基)苯胺可生成菲啶衍生物4u。不对称环丙烯酮反应也表现出良好的区域选择性,给出单一区域异构体4z。
反应机理
基于实验机理研究与文献报道,作者为两条反应路径提出了合理机制框架:
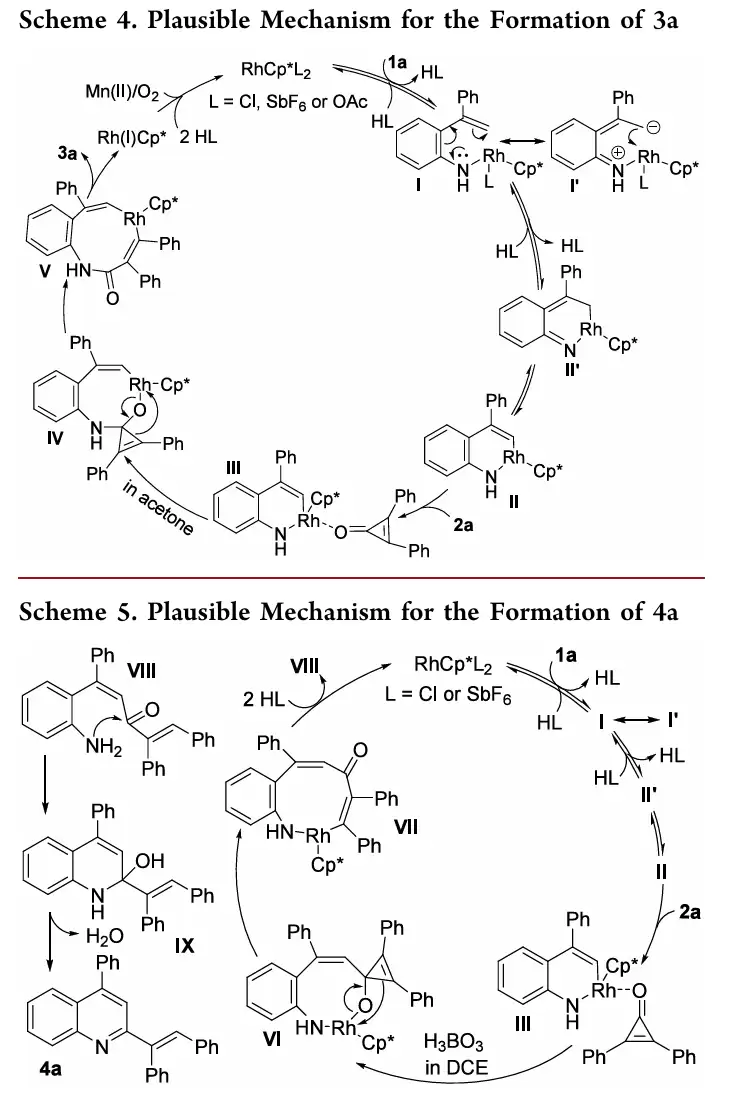
- 合成3a的路径([5+3],条件A/丙酮):Rh(III)与1a发生配体交换生成中间体I,随后通过分子内C-亲核进攻得到II;II与2a配位后形成III,在丙酮溶剂中,2a选择性对N−Rh键发生迁移插入,得到IV;再经β-碳消除和还原消除,最终生成3a,生成的Rh(I)被Mn(OAc)2/O2氧化再生为Rh(III)。
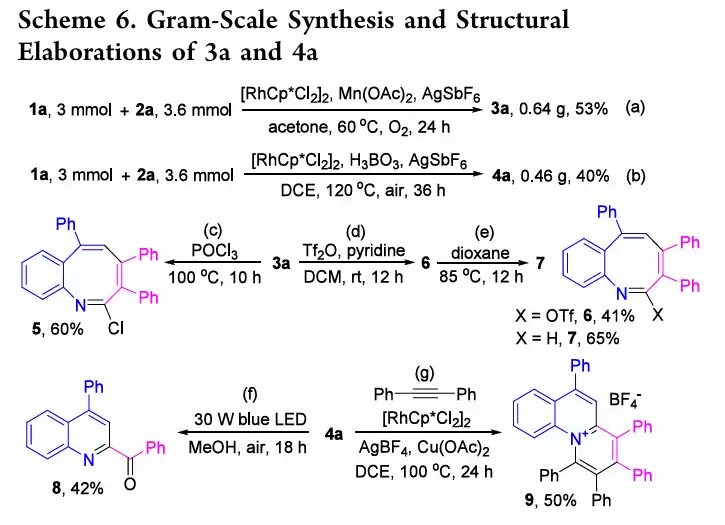
- 合成4a的路径([5+1],条件B/DCE+H3BO3):共享I−III中间体后,在DCE/H3BO3条件下,2a转而选择性对C−Rh键发生迁移插入,得到VI;再经β-碳消除和脱金属质子化,得到VIII和Rh(III);VIII发生分子内N-亲核加成得到IX,最后由芳构化驱动的脱水消除,生成4a。
两条路径的分叉点在于中间体III中2a的插入方式:丙酮环境中优先进行N−Rh键插入(导向3),而DCE/H3BO3环境则优先进行C−Rh键插入(导向4)。这一机理解释清晰明了。
放大合成与产物修饰
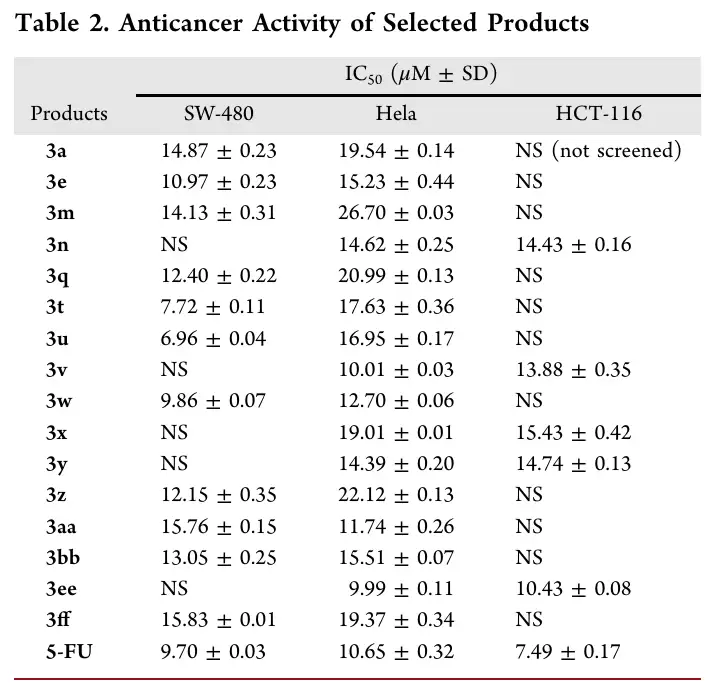
在3 mmol规模下进行放大合成,3a收率为53%(0.64 g),4a收率为40%(0.46 g)。产物还可进一步转化:3a经POCl3处理得到2-氯苯并[b]氮杂辛5;经Tf2O/吡啶及热消除,得到3,4,6-三苯基苯并[b]氮杂辛7。4a在蓝光照射下发生氧化裂解,得到2-苯甲酰喹啉8;与二苯乙炔在Rh(III)催化下反应,得到多环产物9。这些转化实验充分展示了产物的结构拓展能力。
抗癌活性
以5-FU为阳性对照,作者对三种人癌细胞株(SW-480、HCT-116、HeLa)进行了活性评价。结果显示,大多数苯并[b]氮杂辛酮衍生物表现出显著抑制活性,其IC50值与5-FU非常接近,具有进一步研究潜力。具体而言:3u对SW-480的IC50为6.96 μM(5-FU为9.70 μM);3ee对HeLa和HCT-116的IC50分别为9.99 μM和10.43 μM(5-FU分别为10.65 μM和7.49 μM)。而2-烯基喹啉产物4在本次检测中未显示显著细胞毒性活性。
总结
本文发展了一种条件可控的选择性合成策略,通过C−H键活化引发的形式[5+3]或[5+1]环化反应,实现了2-烯基苯胺与环丙烯酮的选择性偶联,高效构建了苯并[b]氮杂辛酮和2-烯基喹啉这两类高价值的N-杂环骨架。该方法底物易得、反应路径新颖独特,所得苯并[b]氮杂辛酮衍生物展现出与阳性对照5-FU相当的显著抗癌活性。此外,反应原子经济性优异,易于放大,未来在相关领域无疑具有广泛的应用前景。