仅12%——这是胰腺癌患者的五年生存率,在所有常见癌症中几乎垫底。三十年来,其背后关键的驱动基因KRAS一直被牢牢贴上“不可成药”的标签。如今,研究人员尝试利用一条肽链,同时按住胰腺癌的两大致命开关。
胰腺癌恶性程度极高,五年生存率仅约12%,其核心驱动基因KRASG12D长期以来被认为是难以成药的靶点;位于下游的HDAC与该基因存在协同作用,但联合用药又常受困于毒性叠加与药物相互作用。近期《J. Med. Chem.》上的一项研究另辟蹊径:通过药效团模型结合分子对接的虚拟筛选方法,从十万余条肽中筛出一条命名为KH-1的肽链,让它用同一条链同时抑制KRASG12D和HDAC——这是该领域首个双靶点肽类抑制剂。本文将带你看清KH-1的设计过程、它一肽两用的分子巧思、从纳摩尔亲和力到细胞与小鼠的完整证据链,以及单药疗效如何反超联合用药;同时也会冷静指出它在HDAC选择性、细胞系基因型与成药性方面尚待回答的问题。读完本文,你将对计算驱动的多靶点药物设计有一个具体而立体地认识。
如果要在所有实体瘤中挑一个最难啃的,胰腺癌大概率名列前茅。它起病隐匿、转移凶猛、对化疗天然耐受,五年生存率长期徘徊在12%左右——这个数字几乎是所有常见癌症中最低的一档。更棘手的是,它背后的核心驱动基因KRAS,曾被业界整整三十年贴着“不可成药”的标签。
最近发表在《J. Med. Chem.》上的这项工作,给出了一个相当新颖的思路:不去做单靶点药物,也不去做联合用药,而是设计一条肽链,让它同时按住KRASG12D和HDAC两个开关。研究者将这个分子命名为KH-1,并声称它是首个同时抑制这两个靶点的肽类抑制剂。
下面我们一层层拆开来看,这个分子是怎么被设计出来的、证据链有多扎实,以及作为读者应该带着哪些保留态度去看待它。
一、先理解:胰腺癌为什么这么难治
胰腺导管腺癌(PDAC)占了全部胰腺肿瘤的约九成。由于缺乏早期诊断的标志物,绝大多数患者确诊时已是中晚期,错过了手术时机。即便能手术,PDAC也以高复发、易转移著称,传统化疗效果有限、毒副作用明显。论文开篇就点明,按照趋势,胰腺癌预计将在2030年成为美国癌症死亡的第二大原因。
在这片荒漠中,KRAS是绕不开的地标。它是一个鸟苷酸结合蛋白,在细胞里扮演分子开关的角色:结合GTP时是“开”(激活下游增殖信号),结合GDP时是“关”。癌症中的KRAS突变集中在三个热点——G12、G13、Q61。其中KRASG12D出现在大约40%的PDAC患者中,是最常见的那一类。
G12D突变破坏GTP水解,让这个开关卡死在“开”的状态,细胞便持续收到增殖指令。问题在于,KRAS蛋白表面光滑,缺少传统小分子能抓住的深口袋,这正是它长期被视为“不可成药”的原因。
二、两个靶点,各有各的硬骨头
这项研究的关键,是它没把宝全压在KRAS一个靶点上,而是引入了第二个:HDAC(组蛋白去乙酰化酶)。理解这两个靶点各自的处境,才能看懂双靶点策略的价值。
KRASG12D这一侧。 尽管难,近年还是有人尝试用环肽去靶向它,比如KD2、KS-58这类分子。但论文直言,这些分子的活性都停留在微摩尔级别(KD2的IC₅₀约12.4 μM,KS-58约30 μM),没有一个能在纳摩尔这种理想的低浓度上结合KRASG12D。后来研究者把KD2改造成KD2-AzaX(文中简称KD2AX),亲和力有改善,但也只到低微摩尔。换句话说,更强的KRASG12D抑制剂,仍是这个领域的待解难题。
HDAC这一侧。 HDAC是一类表观遗传酶,负责把组蛋白上的乙酰基去掉。在癌细胞中,HDAC高表达、异常去乙酰化会促进增殖和血管生成。以Vorinostat(SAHA)为代表的小分子HDAC抑制剂已有不少进入临床,但它们带着血液、胃肠、心脏毒性等副作用,而且在实体瘤中效果普遍不理想——临床上HDAC药物作为单药主要在血液系统肿瘤中有正面结果。
这两个靶点之间还有一层关键联系:HDAC位于KRAS信号通路的下游。已有研究表明,单纯阻断KRAS信号对PDAC疗效有限,而加上HDAC抑制剂后,预后明显改善、能抑制癌细胞自我更新、阻断转移。也就是说,同时打击这两个靶点,理论上具有协同效应。
三、为什么是一个分子,而不是两片药一起吃
既然两个靶点协同,最直接的做法似乎是联合用药——一片KRAS抑制剂加一片HDAC抑制剂。但论文指出,联合用药在临床上有自己的麻烦:剂量依赖的毒性叠加、两种药之间的相互作用、以及药代动力学难以匹配。
于是作者把目标定为:用单一分子同时命中两个靶点,既要获得协同的好处,又要规避单药疗效不足和联合用药的弊端。而且据他们所知,此前从未有人报道过同时靶向KRASG12D和HDAC的抑制剂。这个空白,正是KH-1想填补的位置。
为什么选择肽?相比小分子,肽能覆盖更大的结合界面,对KRAS这种缺乏深口袋的难靶蛋白更友好;相比抗体,肽分子量小、更容易渗透进细胞和组织。代价是肽通常面临稳定性和口服吸收的天然短板——这一点我们在后面评价时再展开。
四、从十万条肽里,筛出五条候选
KH-1不是从实验台上一管一管试出来的,而是先在计算机里筛出来的。研究者采用的是药效团模型加分子对接的组合策略:药效团模型负责快速框定哪些肽具备结合所需的化学特征,分子对接负责更精细地评估结合姿态和打分。这种组合的好处是兼顾了配体与靶点结合时的柔性和动态,比单纯的刚性对接更接近真实。
整个流程像一个漏斗:
- 起点是一个组合生成的2D肽库,共104,976条肽,全部转成3D结构并做能量优化。
- 第一道筛:用KRASG12D药效团模型筛,剩下1807条。
- 第二道筛:再用HDAC2药效团模型筛,剩下51条。
- 第三道筛:把这51条分别对接到KRASG12D(PDB: 6WGN)和HDAC2(PDB: 4LXZ)的活性位点,按对接打分排序,取综合排名最高的5条,命名为KH-1到KH-5。
两个药效团模型怎么来的?研究者基于两个高分辨率晶体结构构建:KRASG12D模型聚焦在Switch II凹槽区域,由两个氢键受体特征和两个氢键供体特征组成,对应Asp69、Arg73、Asp12、Gly60、Gln61、Glu63这些关键残基;HDAC2模型则由两个供体和一个受体特征组成,对应His145、Gly154、Asp181。
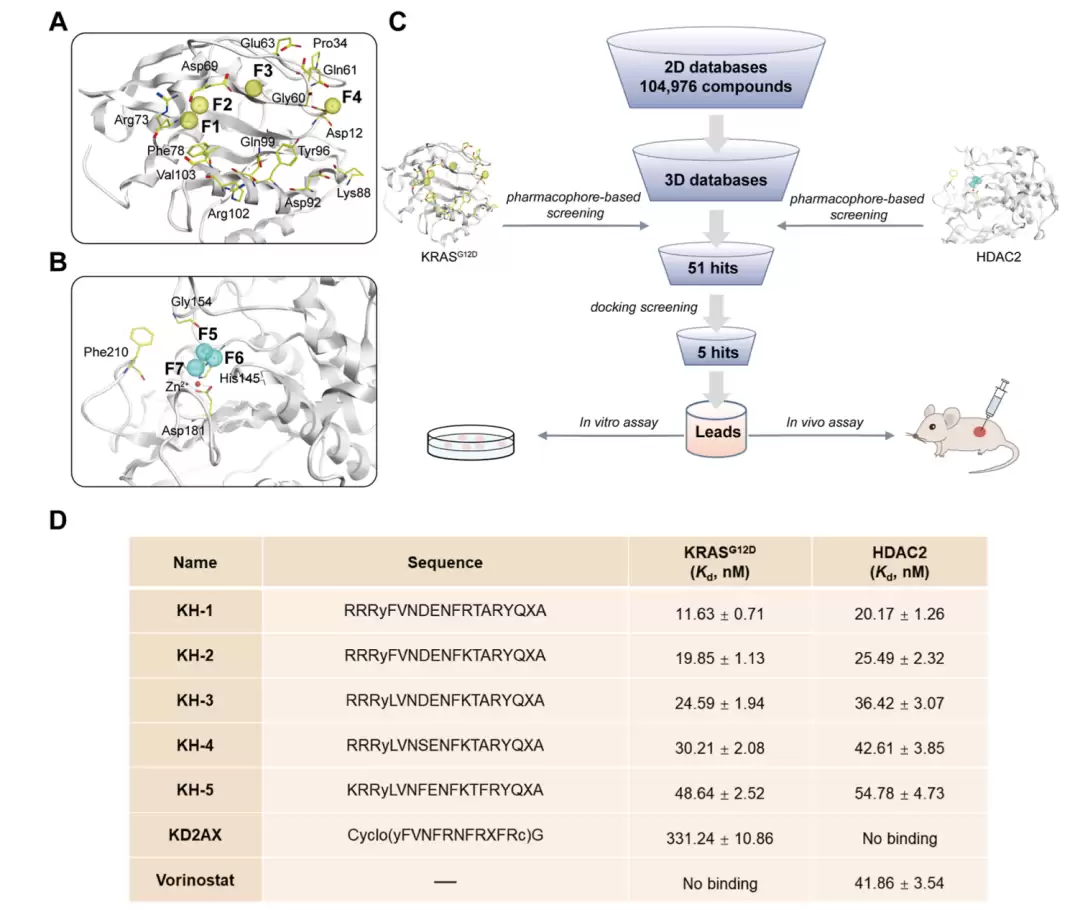
双靶点 KRAS|G12D/HDAC 肽的虚拟筛选。(A) KRAS|G12D 的药效团模型(F1、F2 为氢键受体特征,F3、F4 为氢键供体特征;PDB: 6WGN)。(B) HDAC2 的药效团模型(F5、F6 为供体特征,F7 为受体特征;PDB: 4LXZ)。药效团特征以小球表示,黑色虚线为氢键,靶蛋白活性位点残基以黄色棒状显示。(C) 虚拟筛选流程图。(D) KH 1–5 的序列及其对 KRAS|G12D 与 HDAC2 的结合亲和力;KH 1–5 经 N 端乙酰化与 C 端酰胺化修饰,X 为 2-amino-8-(hydroxyamino)-8-oxooctanoic acid。数据为均值 ± 标准差,n = 3。
值得一提的是计算与实验的吻合度:后续对接打分与实测亲和力之间,KRASG12D的相关系数高达0.978,HDAC2为0.962。这说明他们的虚拟筛选不是碰运气,预测和验证高度一致,KH-1既是打分最高的,也是实测最强的那一条。
五、KH-1长什么样,又是怎么一手抓两个靶点的
KH-1的序列是RRRyFVNDENFRTARYQXA(N端乙酰化、C端酰胺化)。这里有两个设计上的巧思,值得单独拎出来讲。
第一个巧思:那个叫X的非天然氨基酸。 序列末尾的X,是一个人工设计的残基,全名是2-amino-8-(hydroxyamino)-8-oxooctanoic acid。说人话,它是一条八碳长链,末端挂着一个羟肟酸基团。熟悉HDAC抑制剂的读者立刻会反应过来——羟肟酸正是Vorinostat等经典HDAC抑制剂用来咬住活性中心锌离子的“弹头”。研究者等于是把一个SAHA式的锌结合弹头,改造成氨基酸侧链,缝进了这条肽里。结果就是:X残基的侧链伸进HDAC2的锌口袋,与His145、Gly154、Asp181形成氢键,长链则和Phe210发生疏水作用——这一端负责HDAC抑制功能。
第二个巧思:同一个残基身兼两职。 更妙的地方在于,这个X残基(位于第18位)在肽链中的位置,恰好让它直接结合KRASG12D的Asp12——而Asp12正是G12D突变的所在,是区分突变型和野生型的命门。一个残基,一端管HDAC,另一端管KRAS,一肽两用。
除此之外,KH-1的C端ARYQXA这一段,与KRASG12D的Asp12、Gly60、Gln61、Glu63、Lys88形成多个氢键,光是与Asp12侧链就形成了三个氢键;它还与Asp69、Arg73、Asp92、Gln99、Arg102等残基有额外氢键,并与Pro34、Phe78、Tyr96、Val103疏水作用。和已有环肽KD2相比,KH-1多结合了Glu63、Arg73、Arg102几个位点,多出来的这些氢键,被认为是其亲和力更高的原因。
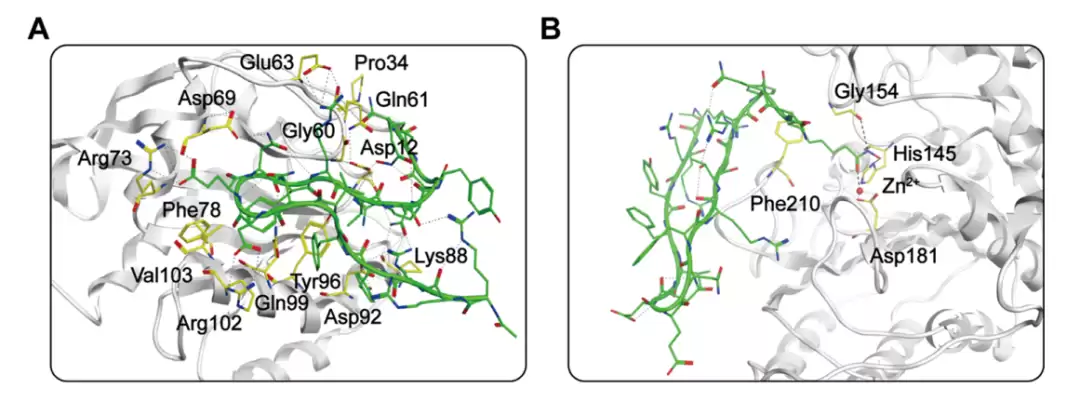
KH-1 的预测结合模式。(A) KH-1 与 KRAS|G12D(PDB: 6WGN)的结合预测。(B) KH-1 与 HDAC2(PDB: 4LXZ)的结合预测。KH-1 以棒状表示(碳为绿色、氮为蓝色、氧为红色),关键残基以黄色棒状显示,黑色虚线为氢键。
研究者还做了细致的构效关系分析,逐个把关键残基突变成丙氨酸,结果突变肽对两个靶点都完全失去结合;随机打乱序列的对照肽同样不结合。这从反面证明了上述这些位点确实是结合所必需的。
六、证据链:从试管、细胞到小鼠
一个分子设计得再漂亮,也要看实验数据撑不撑得住。这篇论文的验证是逐级递进的。
结合力(试管)。 用微量热泳动(MST)测得,KH-1对KRASG12D的解离常数Kd为11.63 nM,对HDAC2为20.17 nM,都达到了纳摩尔级。等温滴定量热(ITC)给出的数值(8.62 nM和17.14 nM)与之基本一致,相互印证。横向比较,KH-1对KRAS的亲和力比KD2AX强约28倍,对HDAC2比Vorinostat强约2倍。
选择性。 这部分有好有坏。好的一面:在KRAS家族中,KH-1只认G12D,对野生型以及G12C、G12R、G12V几乎不结合(Kd都大于10 μM),突变特异性很干净;针对330种激酶的检测也显示几乎没有脱靶。不太理想的一面:在HDAC家族中,KH-1虽然以HDAC2为主(比HDAC1高约45倍、HDAC3约54倍、HDAC6约27倍、HDAC8约18倍),但它对HDAC1、3、6、8仍有微摩尔级的结合。作者自己也承认,HDAC2的选择性不够,是后续优化的重点。
细胞层面。 在两株胰腺癌细胞上,KH-1呈剂量依赖性地诱导凋亡(clea ved caspase-3和clea ved PARP升高)、抑制克隆形成(2.5 μM时几乎不再成团)、降低活力(12.5 μM时抑制率约七到八成)。它把细胞挡在G0/G1期;周期相关蛋白的检测中,cyclin B1随剂量下降、p-cdc2随剂量上升,与细胞无法推进到分裂期的状态相符。它还显著抑制了细胞的迁移和侵袭。重要的是,KH-1对野生型和G12C的胰腺癌细胞、以及正常胰腺上皮细胞几乎没有杀伤(IC₅₀均大于10 μM),显示出一定的选择性窗口。
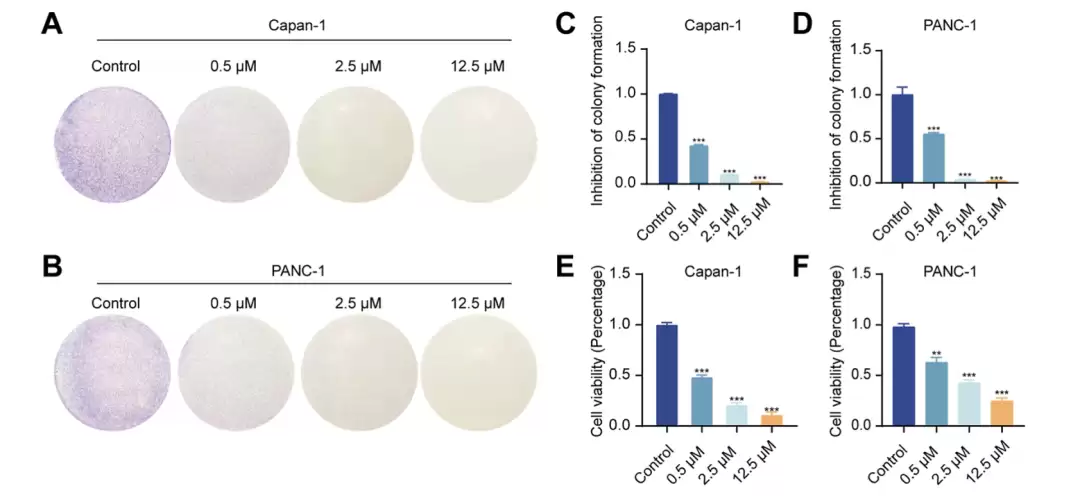
KH-1 对 Capan-1 与 PANC-1 细胞增殖能力的影响。(A–D) 两株细胞经 0、0.5、2.5、12.5 μM KH-1 处理 10 天后的克隆形成代表性图像及定量(C、D)。(E、F) CCK-8 法检测 KH-1 处理 48 小时后的细胞活力。数据为均值 ± 标准差,n = 3;**p < 0.01,***p < 0.001,与对照组比较。
最优雅的一笔:基因敲低实验。 研究者用shRNA分别敲低KRASG12D和HDAC。结果是:对照细胞对KH-1高度敏感(IC₅₀约0.49 μM);单独敲低任一靶点,敏感性都明显下降(IC₅₀升到4到5 μM);同时敲低两个靶点,细胞几乎完全耐药(IC₅₀大于10 μM)。这个梯度漂亮地证明了KH-1的杀伤确实依赖于两个靶点同时存在——这正是双靶点机制最有说服力的证据。
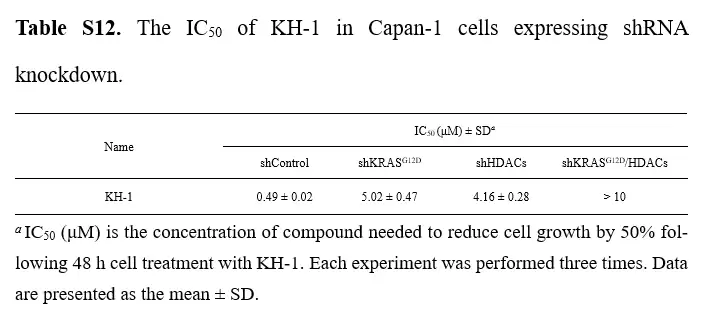
shRNA 敲低 KRAS|G12D 与 HDAC 后,KH-1 对 Capan-1 细胞的 IC₅₀。空载对照(shControl)约 0.49 μM;单独敲低 KRAS|G12D 或 HDAC 后分别升至约 5.02 μM、4.16 μM;同时敲低两者则大于 10 μM。
动物层面。 在胰腺癌移植瘤模型里,1 mg/kg的KH-1显著抑制肿瘤生长,瘤重比对照组低约一半。更关键的对照是:KH-1单药的疗效,超过了KD2AX、Vorinostat各自单用,甚至超过了两者联合用药。组织染色显示KH-1组凋亡标志TUNEL升高、增殖标志Ki67下降。毒性方面,心、脾、肺、肾没有明显损伤,体重、肝肾功能各项血清指标、炎症因子IL-1β和TNF-α都与对照无显著差异。药代方面,腹腔给药后半衰期约8.15小时,血清稳定性良好。
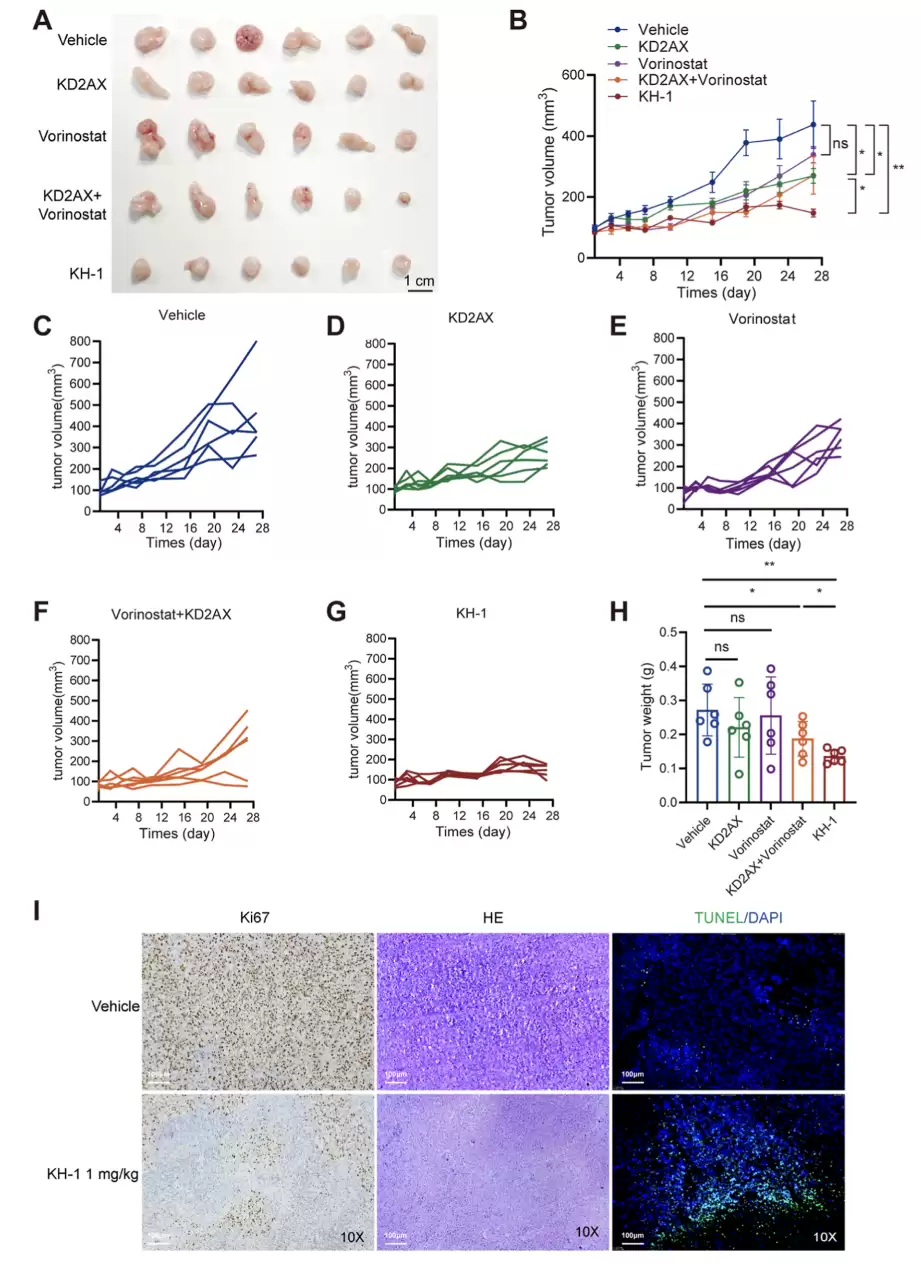
KH-1 的体内抗肿瘤活性。(A) 各组瘤体图像。(B) 各组肿瘤体积变化曲线。(C–G) 空白对照、KD2AX(1 mg/kg)、Vorinostat(1 mg/kg)、Vorinostat/KD2AX 联合组与 KH-1(1 mg/kg)各自的肿瘤体积变化。(H) 各组瘤重。(I) 肿瘤组织 HE 染色,以及 TUNEL/DAPI 与 Ki67 的免疫荧光/免疫组化代表性图像,标尺 100 μm。n = 6;*p < 0.05,**p < 0.01,与对照组比较。
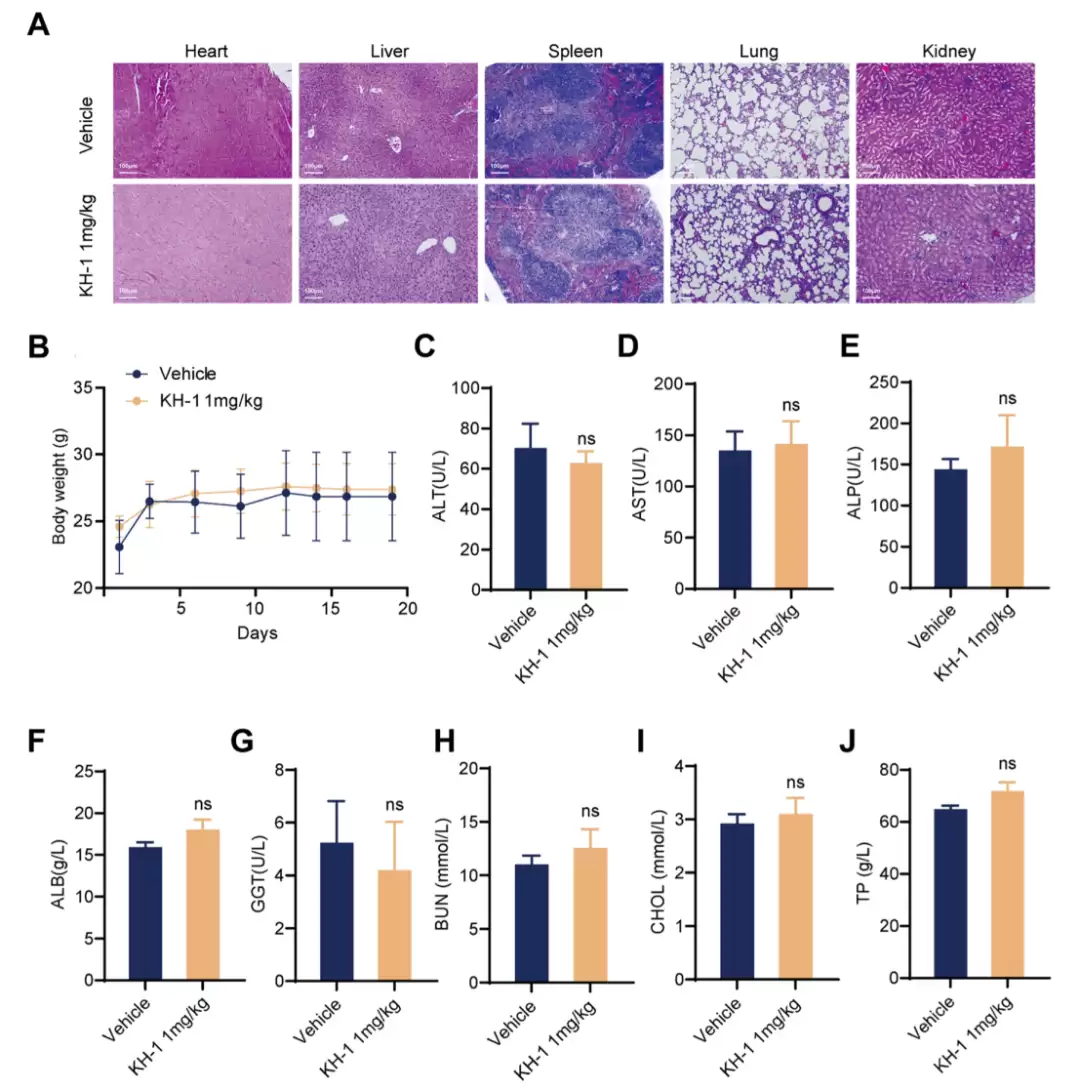
KH-1 的体内毒性评价。(A) 主要器官(心、肝、脾、肺、肾)HE 染色,标尺 100 μm。(B) 小鼠体重变化。(C–J) 血清生化指标(ALT、AST、ALP、ALB、GGT、BUN、CHOL、TP),n = 6。
把这条链串起来看:计算预测、体外结合、细胞功能、机制验证、体内疗效与安全性,方向一致、相互支撑。作为一篇方法学加验证的工作,完成度是相当高的。
七、几个值得回味的细节
反常的剂量关系。 论文提到,KH-1在1 mg/kg的抑瘤效果竟然比10 mg/kg更强,因此后续选用了1 mg/kg。这种低剂量反而更有效的现象并不常见,文中没有给出机制解释。它可能涉及高浓度下的溶解度、聚集、双相药代或免疫相关因素等——无论原因为何,这都是一个值得追问、也值得后续厘清的点。
单药打过联合用药。 一个分子的疗效超过两种成熟药物的联合,这是双靶点策略最想证明的卖点。如果能在更多模型里稳健重现,确实是有分量的结果。
八、怎么评价这项工作:亮点与保留
先说亮点,它们是实打实的:
- 概念新。 首个把KRASG12D和HDAC装进同一条肽的尝试,填补了一个明确的空白。
- 设计巧。 用一个非天然氨基酸的羟肟酸侧链做HDAC弹头,又让同一残基去够KRAS的Asp12,一肽两用,是很聪明的分子设计。
- 计算与实验高度吻合。 0.97量级的相关系数,给这套虚拟筛选流程增加了不少可信度。
- 机制证据干净。 双敲实验把双靶点依赖性讲得明明白白。
- 低剂量有效、安全性初步良好。
再说需要保留和追问的地方——这些不是否定,而是判断一项早期工作成色时该有的审慎:
- HDAC2选择性确实偏弱。 KH-1对HDAC1、3、6、8也有微摩尔结合。这恰恰是临床HDAC抑制剂毒性的来源之一。本研究在小鼠里没观察到明显毒性是好事,但这是预测加短期动物数据,离打消顾虑还很远。作者本人也把它列为头号优化方向。
- 细胞系基因型这一点值得读者特别留意。 论文把Capan-1作为主力细胞系,甚至在Capan-1上做了shKRASG12D敲低来论证G12D依赖性。但Capan-1在ATCC、多项外显子测序与文献中被反复确认携带的是KRAS G12V(纯合)突变,而非G12D(G12D的标准模型通常是PANC-1)。这就带来一个张力:一方面MST数据显示KH-1几乎不结合G12V蛋白,另一方面它在Capan-1里却很有效,而且敲低G12D还能改变敏感性。这中间的逻辑需要更清楚的说明,否则会让人对主力模型与靶点的对应关系存疑。建议读者在引用本文结论时,对这一点保持关注。
- ADMET多为计算预测,并非全部实测。 吸收、分布、代谢、毒性的不少结论来自在线预测平台,尤其肠道吸收被预测为偏弱——这也提示KH-1大概率走注射而非口服。
- 肽类药物的固有挑战未被解决。 递送、体内稳定性、口服生物利用度,这些是肽走向临床的共同关卡。本研究靠腹腔注射给药,在小鼠里跑通了概念,但成药性还有很长的路。
- 免疫原性与长期安全性未知。 这一点作者也在结论里明确承认,需要进一步系统验证。
- 数据来自单一实验室的自存细胞系。 独立机构、更多定义清晰的G12D模型上的重复验证,会让结论更稳。
结语
抛开具体分子,这项工作真正有意思的,是它示范了一种范式:用计算驱动的虚拟筛选,去主动设计多靶点的肽类抑制剂,而不是被动地从天然产物或随机库里淘。把一个锌结合弹头当成氨基酸缝进肽里、再让同一个残基同时够到第二个靶点的命门,这种把化学直觉和计算筛选结合起来的思路,本身就有可复制的价值。
当然,从一条在小鼠身上有效的肽,到一款能真正改变胰腺癌患者命运的药,中间隔着无数道关卡——HDAC选择性、递送、稳定性、免疫原性、人体安全性,每一道都可能让分子止步。KH-1现在的身份,是一个有想法、有初步证据、也有明确短板的早期先导分子。它未必是最终的答案,但它提出问题和解决问题的方式,值得这个领域的人认真看一看。
对一种几乎无药可用的癌症来说,多一个新颖的思路,本身就是好事。





