先把结论放在前面:GalaxyVS 是一套软硬件深度协同的虚拟筛选框架,部署在国家超级计算天津中心。它没有走传统分子对接的老路,而是以 DrugCLIP 的“稠密向量检索”范式为核心,把单靶点对千亿级化合物的筛选,从“数年模拟”压缩到了“数秒检索”,并且首次在十万级靶点×千亿级化合物的规模上完成了系统级筛选,最终产出了一个跨物种的相互作用数据库 GalaxyDB。
研究背景:两个空间的“双重鸿沟”
现代药物发现正同时面对靶点空间与化学空间的快速膨胀,但现有的筛选能力显然没跟上。
先说靶点侧的鸿沟。AlphaFold2 已经为大约 1.58 亿个蛋白预测了结构,覆盖了 UniProt 中约 2 亿条目的 78%。问题在于,那些依赖高分辨率实验结构的传统方法,其搜索空间被死死限制在 PDB 中约 7 万个蛋白上。这占了多少?大约 0.035%。换句话说,绝大多数有高治疗价值的靶点,至今仍是“有结构但没被筛过”的空白地带。
再看化学侧的鸿沟。组合化学让商业可及的化合物库膨胀到了数十亿乃至万亿规模,比如 Enamine REAL Space、WuXi GalaXi、ZINC22 这些。但受限于算力,单次筛选的天花板长期徘徊在约 55 亿化合物的量级,千亿级空间几乎无人敢碰。
经典方法为什么吃力?物理模拟,比如自由能微扰(FEP),精度最高但代价太大,根本没法放大到千亿尺度。分子对接(docking)相对经济,可它还是需要对每个蛋白-配体对反复采样和打分,面对千亿候选时依然力不从心。
深度学习带来了转机。以 DrugCLIP 为代表的方法,把虚拟筛选重构为共享隐空间中的最大内积检索,在效率上对物理模拟形成了代差。同时,大规模合成数据上的预训练让模型对结构变得更鲁棒,可以直接使用 AlphaFold2 预测的结构进行筛选。此前已有工作把人类蛋白质组对 5 亿化合物完成了筛选并公开了数据集。
但从 5 亿到 1000 亿,要跨过三道坎。规模扩大 200 倍绝不是简单“堆量”就能解决的。论文明确指出了三层挑战:
- 硬件层:为 1000 亿分子生成高维稠密向量,所需算力与存储接近 1 PB,远超常规集群的承受能力。
- 软件层:像 FAISS 这样的内存向量检索引擎,需要把整个索引常驻内存。千亿向量即便经过量化,也需要数百 TB 内存,传统方案在此尺度上代价过高。
- 算法层:在千亿库里取 Top 候选,相当于只采样了极小一撮,极易出现多样性坍塌,结果被少数几个骨架的类似物占满。而且快速检索范式本身缺乏精细的物理建模,存在精度损失的风险。
GalaxyVS 正是为同时解决这三层挑战而设计的。

方法内核:DrugCLIP 的稠密检索范式
GalaxyVS 的理论地基是 DrugCLIP——一个多模态对比学习框架,它打破了筛选“吞吐”与“精度”之间的传统取舍。
双编码器与对比对齐
DrugCLIP 使用两个独立的、基于 Uni-Mol 的 Transformer 编码器,分别处理小分子与蛋白口袋的 3D 原子特征。分子编码器加载了预训练的 Uni-Mol 权重,口袋编码器则在 ProFSA 数据集上通过对比蒸馏与之对齐。两者通过对比学习,把匹配的蛋白-配体对在表征空间中拉近,把不匹配的推远。
为什么这能解锁千亿尺度?
关键在于双编码器是完全解耦的:分子编码与蛋白完全独立。这意味着你可以把整个化合物库离线一次性编码并落盘。每次新的筛选,只需在线编码目标口袋,再计算它与全部预计算分子向量的余弦相似度,然后排序。于是,几何依赖的亲和力估计被改写为高效的向量内积运算——这正是把“在线检索”从“离线准备”中剥离出来的结构性前提。
集成与分数归一化
为了增强对构象变化的鲁棒性,每个分子的最终向量由 6 个交叉验证模型集成得到。多口袋构象下的原始余弦分数,则采用调整后的鲁棒 Z-score 归一化以保证可比性。每个分子的最终分数,取其在所有相关口袋上归一化分数的最大值。
系统架构:三大技术支柱
GalaxyVS 并非现有工具的简单拼接,而是从底层硬件到顶层候选精炼的系统性重构,由三个相互衔接的模块组成。
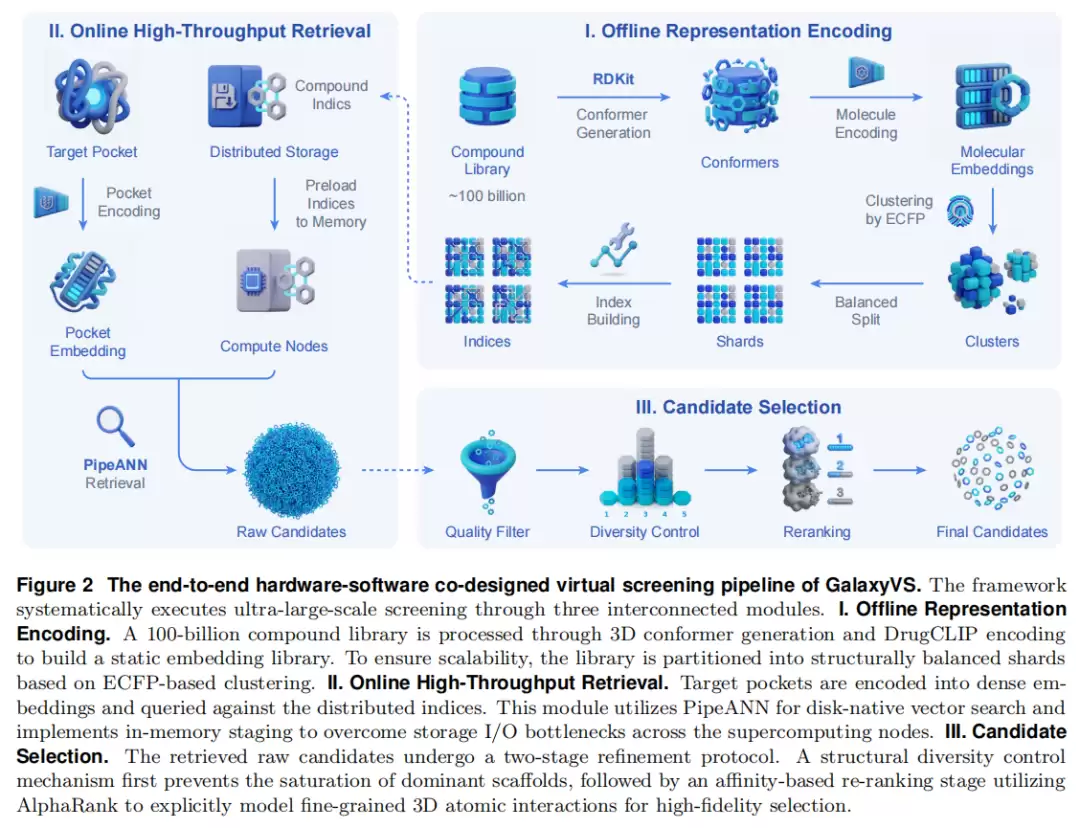
模块一:异构架构上的大规模表征编码
流程上,两个库都以 SMILES 提供,先用 RDKit 为每个分子生成低能量 3D 构象,再送入 DrugCLIP 分子编码器抽取静态向量。落盘后的向量库规模约 200 TB。
硬件适配方面,团队把 DrugCLIP 工作流从 CUDA 迁移到了天河超算的异构翻跟斗上。他们将计算核重写为设备相关算子,并通过统一算子接口注册,在保持 PyTorch 编程模型的同时实现了透明分派。针对 Transformer,他们实现了融合多头注意力算子,把线性投影、矩阵乘、softmax 与 dropout 合并为单个 kernel,减少了启动开销,提升了片上数据复用。
调度与容错方面,设计了节点感知的任务分配策略与失败重提机制,以在长时间编码作业中维持吞吐稳定。
离线成本,也就是一次性沉没成本:构建这套千亿库消耗了 1000 个 CPU 节点×12 天,以及 5000 个异构翻跟斗节点×18 天。这正是 GalaxyVS 吞吐飞跃的关键——把构象生成、向量编码这些重活全部前置为离线一次性投入,在线检索则近乎瞬时。
平台一致性验证:为了确认翻跟斗与原始 GPU(NVIDIA A100 80GB)数值一致,团队抽样了 100 万分子在双平台上编码。结果显示,逐元素差异在小数点后第 4–5 位,均值 6.86×10⁻⁵,最大 5.75×10⁻³。在 744 个构象、53 个口袋组上的排序一致性(Spearman)达到 5–6 位有效数字,Top 1% 差异仅数十个分子,处于实践可接受的范围。
模块二:高通量向量检索(PipeANN + 双模式)
磁盘原生索引(PipeANN):为了避免数百 TB 的内存常驻,团队引入了 PipeANN。它的索引结构与 DiskANN 相同:向量组织为有向图,图索引存盘以大幅压低内存占用,仅把 PQ 压缩向量驻留内存以加速访问。索引遍历采用“偏置最优优先搜索”,反复发起 4KB 随机读直至收敛。
I/O 工程细节:索引文件存放在基于 HDD 的 Lustre 并行文件系统上。为了掩盖随机读的高延迟,PipeANN 用最大 I/O 深度 32 的自适应流水线,重叠计算与磁盘 I/O,并利用多盘并行。这套磁盘原生方案,构成了下文“亲民模式”的基础。
关键配置:每个分区约 100 万向量(共约 10 万个分区);多数分区图最大出度 R=64,连通性弱的“困难分区”提升至 128;构建时候选池 L 默认 100、大 R 时 192,检索时 L=5×top-K;每个 PQ 压缩向量 32 字节,内存-磁盘比 1:128(全集群 <10 TB 内存)。
两种运行模式:
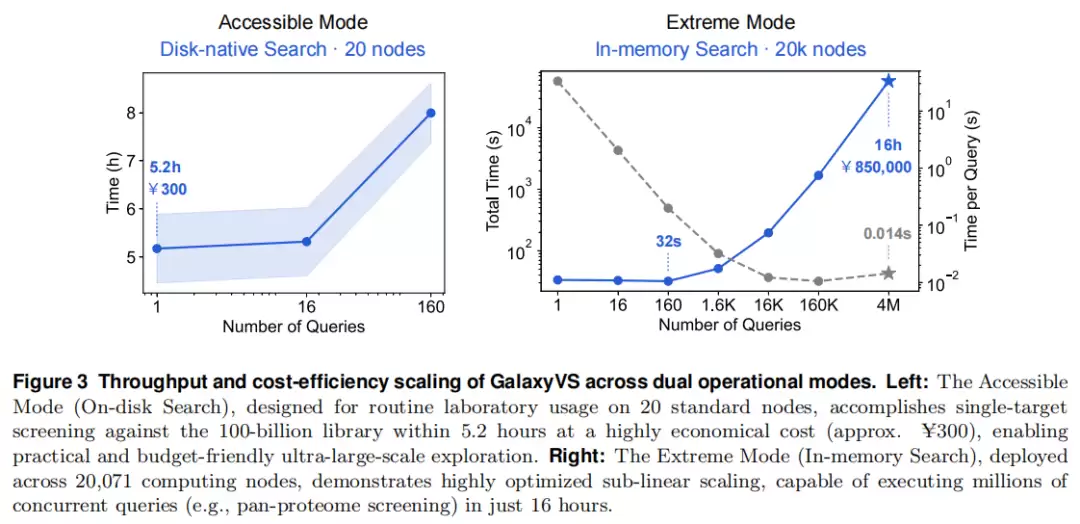
| 模式 | 硬件 | 任务 | 性能 | 成本 |
|---|---|---|---|---|
| Accessible(亲民) | 20 个标准节点 / 磁盘检索 | 单靶点 | 5.2 小时 | ≈ ¥300 / 靶点 |
| Extreme(狂飙) | 20,071 个节点 / 内存检索 | 全蛋白质组 | 160 口袋 32 秒;日吞吐 1.5×10¹⁶ | 大规模算力(图示约 ¥85 万级) |
“狂飙模式”把磁盘检索升级为分布式内存检索:将分片预加载进各节点本地内存,使检索摆脱共享存储 I/O 瓶颈。它呈现出明显的亚线性扩展——当并发查询冲到百万级时,单口袋有效延迟低至约 0.014 秒。
模块三:多样性控制与亲和力重排序
检索得到的原始候选先经鲁棒 Z-score 归一化,随后进入“两阶段精炼”,以同时保证化学多样性与排序精度。
(a) 结构感知的分区:在建索引前,用 K-Means + ECFP4 指纹把全库聚成约 1 万个结构家族。因簇大小极不均衡,再均匀细分为约 10 万个大小相近的结构内聚分片(每片约 4–40 百万分子)。
(b) 多样性因子:检索 Top 0.01% 后约得 1000 万分子。直接取全局 Top 会导致扎堆,因此采用两阶段选择:先从每个分区独立取 Top 比例 r,再从并集中选最终 top-k。随着 r 增大,最终被代表的簇数 l 单调上升至 min(k, 10000)。给定目标 k 与期望 l,用二分搜索高效确定最优 r——只需对 1000 万级集合做一次全局排序,分钟级即可完成。
(c) 性质与规则过滤:全库不预过滤(保留灵活性),而是对每个检索子集按需过滤。单靶点 ABFE 评估采用较宽松规则:
| 性质 | 取值范围 | 性质 | 取值范围 |
|---|---|---|---|
| 分子量 | [150, 550] | TPSA | [0, 200] |
| 环数 | [1, 7] | 可旋转键 | [0, 12] |
| H 键供体 | [0, 6] | 芳香环数 | [1, 7] |
| H 键受体 | [0, 12] | 最大环尺寸 | [3, 8] |
| ClogP | [−3, 5] | 异构体数 | [1, 8] |
同时,排除 PAINS、ZINC 警示结构及多醚酯、双胍、硝基等模式;允许原子类型限定为 {H, C, N, O, F, Cl, Br, I, S, P}。
(d) AlphaRank 重排序:多样化候选随后由 AlphaRank(构建于 AlphaFold3 骨架,以成对排序损失优化)做精细亲和力重排序。与检索阶段粗粒度的语义向量不同,AlphaRank 显式建模口袋-配体的 3D 原子级相互作用与空间构象,把“数学上的近邻”精炼为“生化上严谨的候选”。
实验结果
速度与吞吐:把计算瓶颈“转移”掉
亲民模式以 20 节点、5.2 小时、约 ¥300 完成单靶点千亿筛选。狂飙模式在 20,071 节点上 32 秒完成 160 口袋批量检索,满负荷日吞吐达 1.5×10¹⁶ 次打分——相对于此前 docking 类超算纪录(SWDOCKP2)提升约百万倍。其本质是范式转变:把重活前置为离线沉没成本,在线只剩近乎瞬时的向量运算,从而实现亚线性扩展。
化学多样性
在 102 个 DUD-E 靶点上,与一个聚合自 ChemDiv、ChemBridge、Enamine、Life Chemicals 的代表性“在库”基线(过滤后 2.94M)对比:基线取每靶 Top 30,000(约 1%);千亿库取 Top 0.01% 后经多样性控制保留 30,000 个、覆盖约 8,000 个簇。
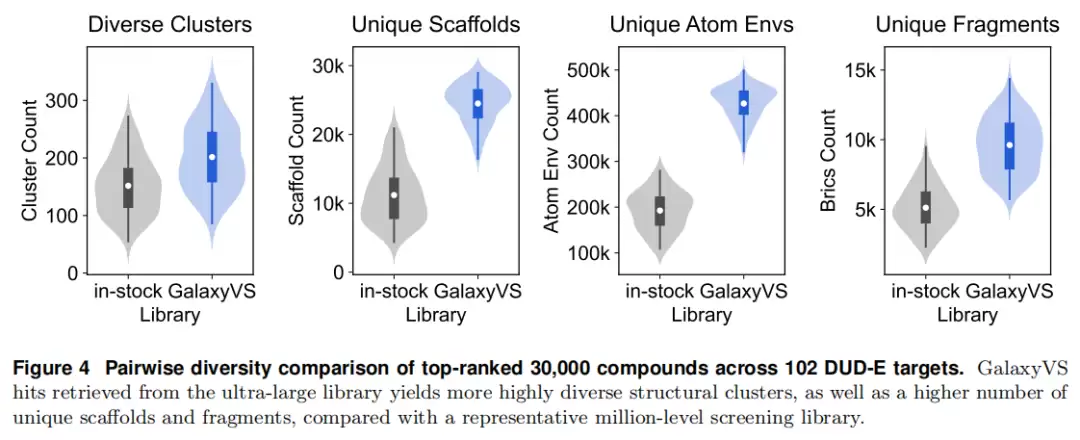
评估采用 Leader–Follower 聚类(ECFP 1024-bit,阈值 0.85)、Bemis–Murcko 骨架、Morgan 半径-4 原子环境、BRICS 片段四类指标。结果显示,GalaxyVS 命中的分子在独特簇、独特骨架、独特原子环境、独特片段上均显著高于百万级库,有效缓解了常规流程中的结构冗余。
化学新颖性
针对 42 个在两种库中均存在新颖性挑战的靶点,基于 PubChem 把命中分子分为四级新颖度:高(未收录且 <5 类似物)、中(已收录且 <5,或未收录且 ≥5)、低(已收录且 ≥5)、最低(专利覆盖)。
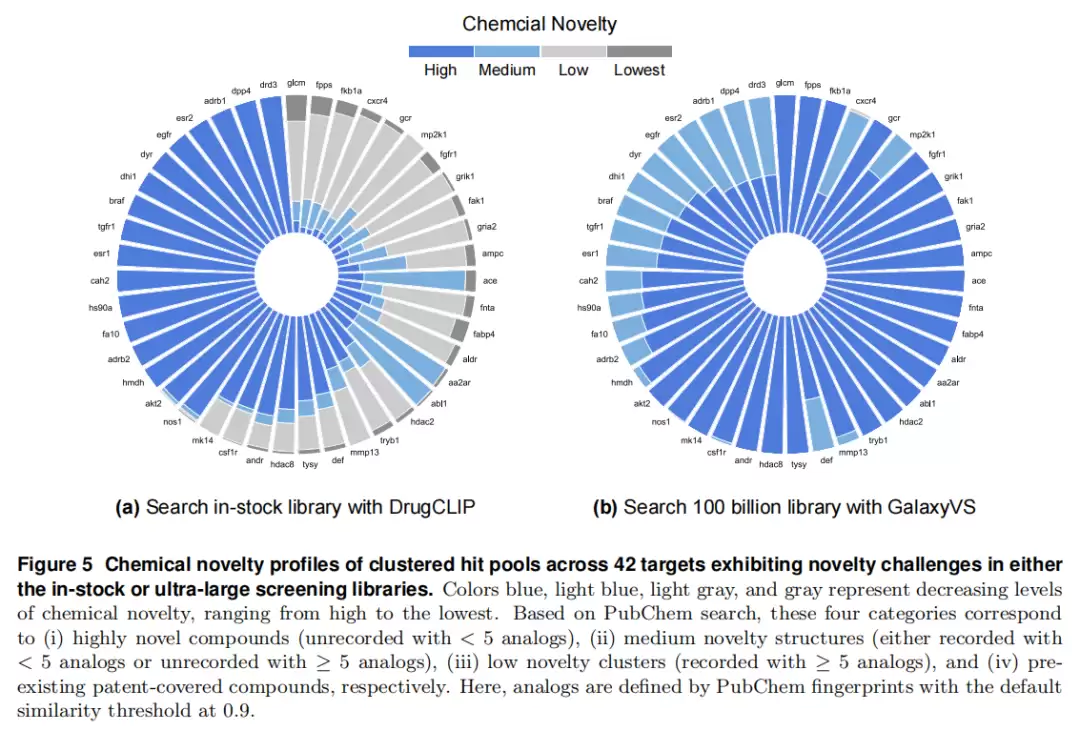
对比鲜明:传统在库筛选产出大量“低/最低新颖度”分子,集中在拥挤且可能受专利约束的区域。而 GalaxyVS 即使面对最难靶点,命中也主要落在中等新颖度区间——更可能是尚未报道、仅与已知分子部分子结构相似的全新结构。
预测亲和力:打分 + 自由能双重验证
Boltz-2 打分:对每靶用簇代表选取至多 60 个化合物。千亿库的 Boltz-2 分数分布明显向更强亲和力偏移,整体胜率 80.4%。
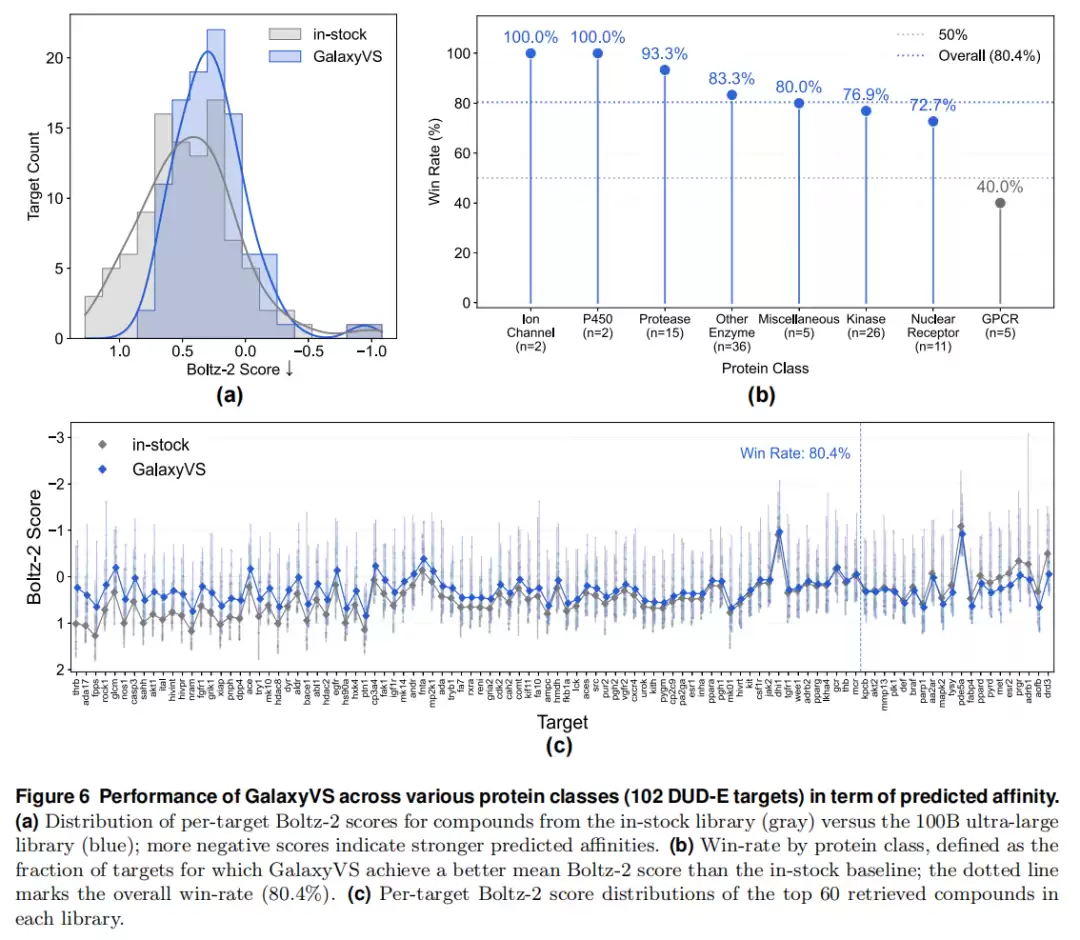
| 蛋白类别 | 胜率 | 样本数 | 蛋白类别 | 胜率 | 样本数 |
|---|---|---|---|---|---|
| 离子通道 | 100.0% | 2 | 其他酶 | 83.3% | 36 |
| P450 | 100.0% | 2 | 激酶 | 76.9% | 26 |
| 蛋白酶 | 93.3% | 15 | 核受体 | 72.7% | 11 |
| 杂项 | 80.0% | 5 | GPCR | 40.0% | 5 |
酶类与核受体普遍 >70%。GPCR 仅 40% 是唯一明显偏低项——这与 GPCR 高度构象柔性、静态打分函数难以区分真实结合者与背景噪声的已知难点一致(且 n=5 样本偏小)。
ABFEP 自由能验证:以 BRD4 为靶,用 BAT.py 对 Top 10 分子做绝对结合自由能微扰:平均 ΔG = −6.25 kcal/mol、标准差 1.57,平均统计误差 1.12,收敛良好。其中 GVS-001 ΔG = −8.97 ± 0.89、GVS-002 ΔG = −8.03 ± 0.85 kcal/mol;二者采取经典结合模式,与 ASP381、关键锚定残基 ASN433 及保守水分子形成稳定相互作用网络。
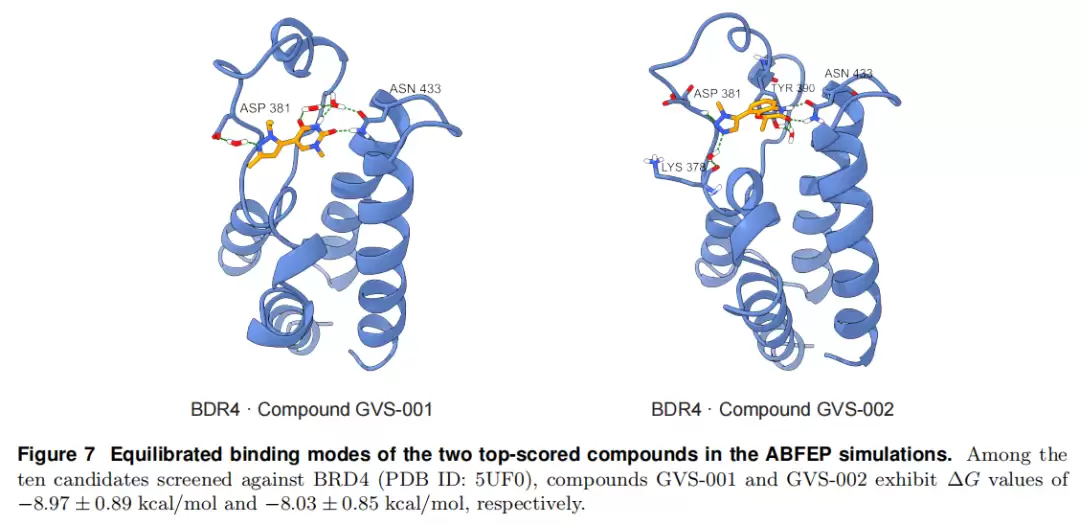
与 docking 选择的对比:按 docking 分数选的候选能找到更低的表观能量极小(如 Compound 16 ΔG = −10.97),但波动更大——Compounds 11、13 出现弱结合(ΔG ≥ −3.00)且标准差高达 2.98,提示在显式溶剂中结构不稳、部分脱离口袋。相比之下,GalaxyVS 更倾向热力学一致、收敛良好的分子。
检索精度与极端富集
PipeANN 精度:检索时 top-K=100、L=500,随机抽三个索引对照穷举基线,平均召回 97.67%(98/97/98)。论文认为,早期筛选阶段 <5% 的遗漏可以接受,因为初筛只是粗滤,后续还有结构方法精算。
极端富集:在 0.01% 的极窄检索深度下,GalaxyVS 于 DUD-E 取得 EF₀.₀₁% = 1594.3,LIT-PCBA 取得 297.6,表明其在超大背景与极窄保留比下仍保有强富集力。
跨物种筛选与 GalaxyDB
凭借高吞吐与对 AlphaFold 结构的适配,团队把筛选从单靶点扩展到了整个蛋白质组,选取了 6 个跨演化分支的代表性物种,结构数据取自 AlphaFold 数据库:
| 物种 | 类型 | 结构数 |
|---|---|---|
| 人类(Homo sapiens) | 哺乳动物 | 23,586 |
| 小鼠(Mus musculus) | 哺乳动物 | 21,452 |
| 拟南芥(Arabidopsis thaliana) | 植物 | 27,402 |
| 果蝇(Drosophila melanogaster) | 昆虫 | 13,461 |
| 酿酒酵母(S. cerevisiae) | 真菌 | 6,055 |
| 大肠杆菌(E. coli) | 原核 | 4,370 |
| 合计 | 96,326 |
筛选规模:经口袋识别与结构过滤后,筛选空间约 400 万个结合构象、来自约 10 万个蛋白靶点。整场战役在 20,071 节点上 16 小时内完成,累计 4.0×10¹⁷ 次口袋-配体打分,单口袋有效延迟约 0.01 秒。
GalaxyDB 构建:经 elbow 分析在 102 DUD-E 靶点上确定目标多样性约 7,000 簇。每个百万级富集子集在 z-score < −4 约束下保留 30,000 分子并最大化簇覆盖,再以 Leader–Follower 聚类取代表。随后用 AutoDock Vina v1.2.5 做集成对接:仅人类数据集即覆盖 26,562 个口袋、187,715 个口袋构象,每个口袋约 200 个候选配体、exhaustiveness=16,保留最优 pose 用于建库。
这份跨物种相互作用全景图将开放释出,可支撑人类疾病治疗、抗菌发现、农药/除草剂开发等多个方向。
化合物库与评测基准
两大库合计约 94.00B,也就是常说的“1000 亿级”:
- Enamine REAL Space(约 64.86B):源自 181,288 个合格砌块、172 套合成方案,所有分子 ≤3 步可合成,成功率 >80%、交付 3–4 周(采用 2024 年 7 月版)。
- WuXi GalaXi(29.13B):整合 >30 种可并行反应与药明的砌块/骨架库,约束 ≤3 步、可行性 60–80%、交付 4–8 周(采用 2025 年 1 月版)。
与既有大规模筛选平台的对比:
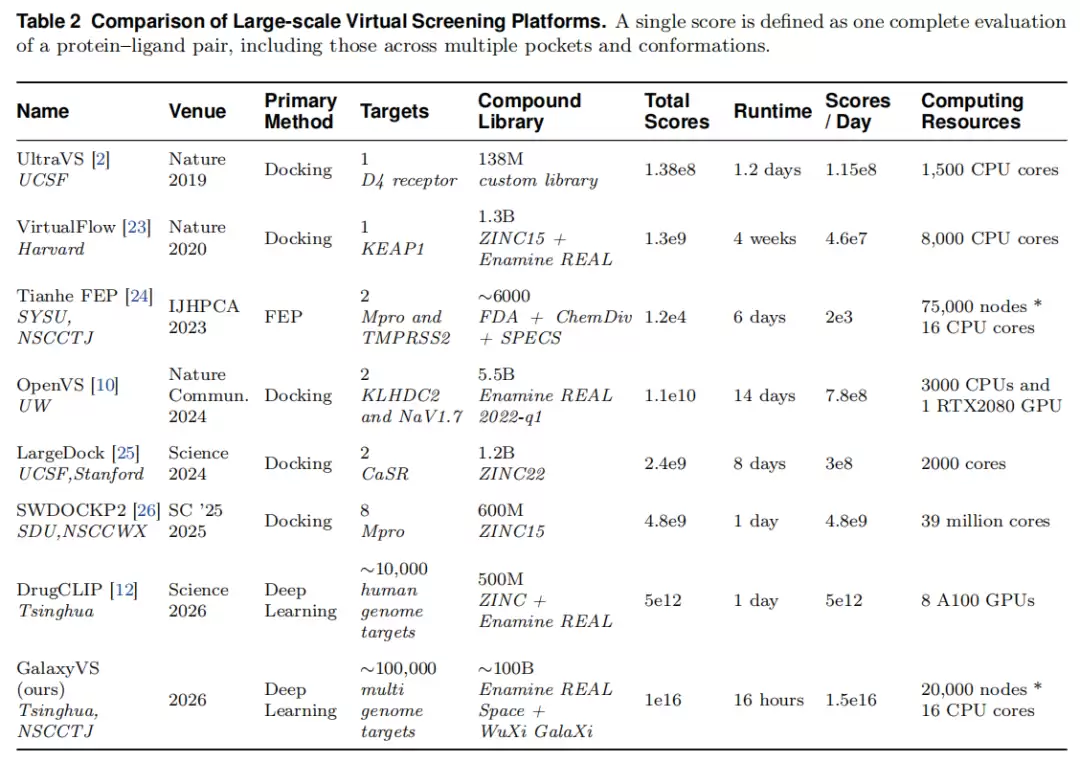
基准:DUD-E(102 靶,平均 224 活性 + 约 62 倍诱饵)评估跨家族富集;LIT-PCBA(14 靶、7,761 活性、382,674 实测非活性)以实验数据替代诱饵,更贴近真实但活性比例差异大。
亮点、局限与展望
主要贡献
- 范式落地的工程闭环:不止提出方法,而是从底层算子、磁盘原生索引到候选精炼做了完整的系统级工程,并在国家级超算上真实跑通。
- 可及性与极致吞吐双模式:¥300/靶的亲民模式让千亿筛选“平民化”,狂飙模式则支撑全蛋白质组级扫描。
- 质量未被速度牺牲:多样性、新颖性、预测亲和力三方面均较百万级库有可量化提升,并用 ABFEP 做了物理验证。
- 开放数据资产:GalaxyDB 作为跨物种相互作用图谱开放释出,具备社区价值。
局限与需审慎之处
- 尚未经同行评议,且全部验证为 in silico(Boltz-2、AlphaRank、ABFEP),缺乏湿实验确证——论文亦将其列为未来工作。
- 评估模型的同源性:Boltz-2 与 AlphaRank 同以 AlphaFold3 为骨架,用一族相关模型做“重排序”与“评估”可能存在一定循环性,理想情况下需独立实验背书。
- GPCR 等柔性靶点偏弱(胜率 40%),静态打分对高构象柔性靶点的固有短板依然存在。
- “百万倍”是吞吐口径的跨范式比较,并非同口径逐步提速。
- 检索为近似:PipeANN ~97.67% 召回意味着约 2–3% 的真实近邻被漏掉。作为初筛可以接受,但属于需要知晓的取舍。
- “1000 亿”实为约 940 亿,且为两库的枚举子集而非全空间;DUD-E 诱饵为计算生成,未必反映真实库分布。
- “亲民”主要指亲民模式;跨物种战役本身依赖 2 万节点的大规模算力,并非低成本。
展望
随着 AlphaFold 结构覆盖与可及化学库继续扩张,“检索式”筛选有望成为常态化的一线工具。后续如果能补上系统性湿实验验证,并将 GalaxyDB 与下游优化、ADMET、可成药性评估打通,其对真实管线的价值将更为可观。




