编者按
材料研发的“试错时代”,正在被AI加速改变。5月21日,未来光锥「AI for Science 创变者说」第二期沙龙“AI+材料的千亿级机会”,邀请了三位学界与产业一线嘉宾,共同探讨AI+材料科学的前沿与实践。北京中关村学院首席材料科学家、开物纪创始人陆子恒,正带领团队用AI重新定义材料研发,直播中,他分享了理想的材料模型长什么样,团队如何暴力穷举一个关于新材料的答案等问题。
材料科学领域的每一次跨越,几乎都对应着一个文明时代的名字——石器、青铜、铁器,再到今天的硅基时代,这么说一点也不夸张。一个新材料的诞生,远不止是实验室里多了一个样品,它往往能撬动一整条产业链,甚至改写人类文明的进程。
高效地设计出一种拥有指定性能的新材料,是材料科学家们长久以来的梦想。过去一两百年里,我们确实收获了不少重磅成果,比如硅、钙钛矿等等。但追根溯源,找新材料始终绕不开两类基本问题:
其一,能不能更快地找到那种“更高、更快、更强”的材料?
举个例子,是否存在一种材料,它的导热能力比金刚石还强,能把芯片运行时产生的热量高效散发出去?这个问题一旦解决,对芯片行业的贡献将是碘伏性的。但难度也摆在眼前:元素周期表里有一百多种元素,它们能组合出近乎无穷无尽的新材料,这个搜索空间实在太大了。
其二,更本质也更尖锐——这种材料到底存不存在?
比如,在常温常压下,到底存不存在电子超导体?这个问题决定了我们是否还要投入一代又一代科学家的青春,去寻找那个“不知道是否存在”的答案。在过去,没有AI,我们面对这个问题几乎束手无策。根本原因仍然在于那个天文数字般的搜索空间,以及过于漫长的统计物理计算链路。
拿碳来举例。一个稳定分子或晶体,其最基本的要求是结构落在一个“能量极小点”上。下图展示了随着碳原子数量增加,可能的能量极小点数量的变化——这是一个典型的超指数增长曲线。一旦原子数超过10个,数字庞大到完全超出想象。也正是因此,解答那种“存在性”问题才变得异常艰难。
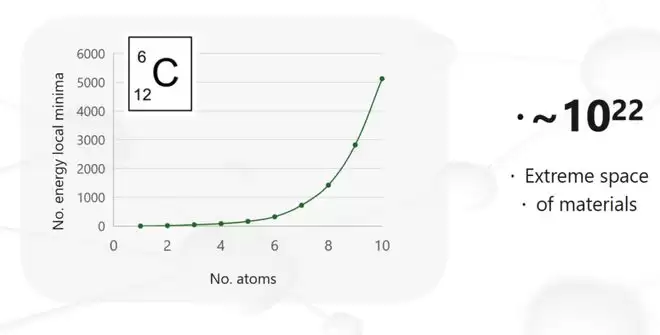
不过,回过头看过去三五年,人工智能恰好给了我们一个绝佳的突破口。无论是AlphaFold还是AlphaGo,这类AI模型最擅长的,恰恰就是处理这种“指数爆炸”问题。
理想中的材料模型
理想情况下,要解决上述两个问题,材料模型应该长什么样?
第一,随便给一种材料,能不能不做实验,就快速、准确、可泛化地预测出它的属性?这样一来,相当于把材料筛选的速度提高了上千倍,寻找有价值的材料自然更快。
第二,万一自己拍不出钴酸锂这种厉害的材料怎么办?有没有可能做一个AI模型,我给定一个目标性质,它就能快速把符合条件的材料或小分子找出来?这样就不再完全依赖人类的灵感了。
先从正向预测说起。在微软期间,我们开发了MatterSim这个模型。它的任务是:给定任意一种材料,用一个大模型,快速、可泛化、尽量不依赖实验地获知其性质。
(图片来源:https://github.com/microsoft/mattersim/)

要建成这样的AI,最核心的能力是泛化性。原因很简单:我们在寻找新材料时,并不知道好材料究竟藏在哪个角落。那些真正重磅的材料,往往都是训练数据里从未出现过的。理想的材料模型,需要对任意元素组合、任意温度、任意人类可控压力下“拍脑袋”想出的材料,都能给出靠谱的预测。
那么,这个目标有理论依据吗?原则上,只要知道一种材料在原子级别的基本性质——比如能量、力、波恩有效电荷、磁矩等等——再配上统计物理的手段,就能推导出所有宏观性质。过去,我们真的尝试过这样做,但非常困难。要拿到这些微观性质,必须解薛定谔方程,实际上解的是简化后的密度泛函理论方程。方程本身能解,但问题在于:要得到宏观性质,你需要做统计平均、热力学平均,进行大量采样。计算量巨大,很多时候根本算不动;就算硬算,也做了大量简化,精度大打折扣。
那么,有没有可能用AI把这件事办了?
思路听起来很简单:给定一个结构,原来解方程拿物理性质,现在换成神经网络直接预测。这本质上就是一个有监督学习的问题。但这个思路有一个“bug”——还是泛化性。我要找新材料,可我并不知道新材料长什么样,那就得先做数据、训练一个模型,再拿它去找——这不就成了一个死循环吗?
所以,我们需要的是一个通用到近乎“蛮横”的数据集——不是在某个组分、某个温度、某个压力下能预测,而是在任意元素组合、任意温度、任意人类可控压力下,都能接住我随便“拍脑门”想出来的材料。
下一个问题是,数据集怎么造?
我们采用了一套比较朴素但有效的方法:主动学习加局部采样。简单说,就是让机器自己去挑选那些“最具代表性”的数据,然后再把这些数据喂给方程去求解、收集结果。这样做下来,对数据量的需求降到了原来的百分之一左右。
即便如此,数据量的绝对值依然非常吓人。当时可以公布的大概是1700万条,最近新挂出来的一篇arXiv论文里已经做到了3500万条——实际上每年还在以近亿的规模往上涨。这就是MatterSim的底气。
(来源:https://arxiv.org/abs/2605.07927v1)
当给定MatterSim一个材料结构,它可以零样本推断出最基本的物理量——能量、力、应力。除此之外,它还能处理电、磁、光等其他模态的性质。
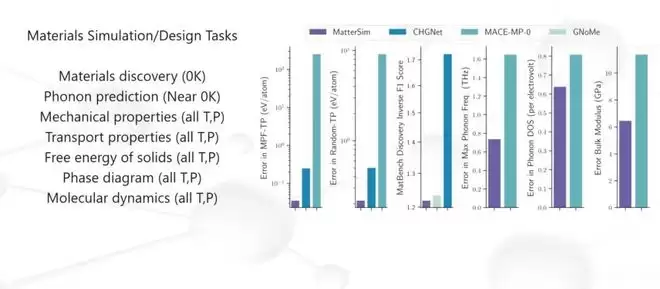
有了这个“硅片上实验”的平台,材料科学家不再需要每次都在炉子前苦等结果。他们可以在计算机里先跑一遍“干实验”,把候选材料从海量可能性中快速过滤出来,再交给真实的“湿实验”去验证。这就是MatterSim的核心价值:用AI把材料研发的“试错”变成“筛选”。
于是,一个价值十亿美元的问题可能到了可以触及的程度。
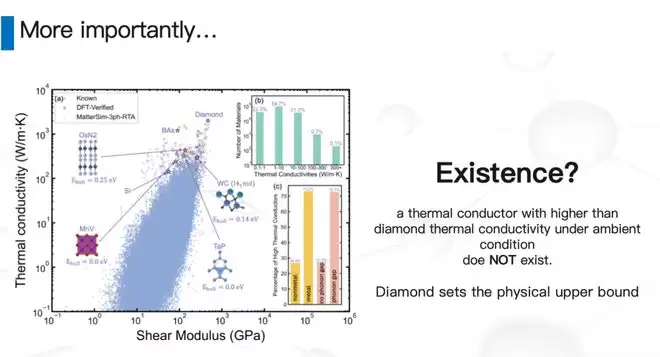
19世纪,人类就发现金刚石热导率很高,而到今天为止,体相材料里基本没有热导率能高于它的。所以一直有个疑问:金刚石是不是就是热导率的上限了?热导率被锁死了吗?
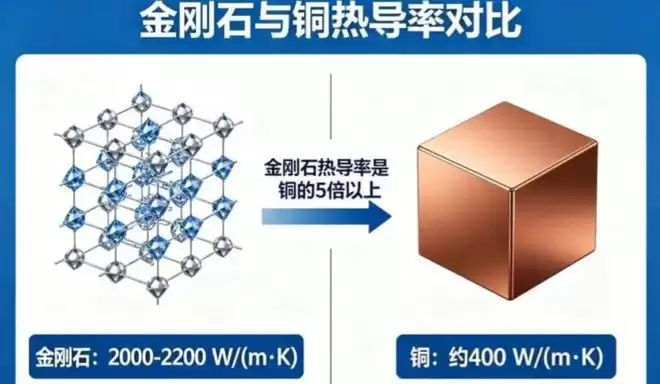
基于此,我们用MatterSim去解答这个问题。
刚才提到,原则上我们可以从微观性质+统计平均拿到材料的宏观性质。对散热来说,最关键的过程叫声子散射。过去要做这种统计计算非常慢,但有了MatterSim这种模型之后,计算速度大大提高——因为模型如此之大,精度也非常之高。
原来计算一个晶体热导率要好几周,现在我们能把速度提高到原来的上万倍,并且精度有保证:几分钟、30秒就把热导率做出来,误差还比较小。
有了这个东西,我们就干了一件非常暴力的事。
我们把元素周期表里:所有一元体系、所有任意两个元素的组合、一部分三元素的组合,以及各种化学计量比和各种空间几何结构的组合,做了一个暴力穷举。
做了很久,花了很多钱,最终得到了几个结论。我们大概有23-24万个结构“有那么一丁点儿没合成过、且有热导率值得算”的概率,每个材料的热导率我们都算了出来。
结论一:热导率高的材料比例非常低。注意,这是log scale——热导率高于1000的非常稀疏。这也是为什么这么多年来找高热导率材料这么慢,因为实在太稀疏了。
结论二:虽然稀疏,但因为我们做了非常大的穷举,依然能找到一些非常有意思的候选材料。最近有一个TaP、一个TaN,一个是我们找合作伙伴一起合出来了,另一个TaN是UCLA的一个教授合出来的,热导率预估和我们基本接近——这是个非常好的信号,确实合成出了一些热导率比较高的、有可能帮我们做芯片散热的新候选材料。
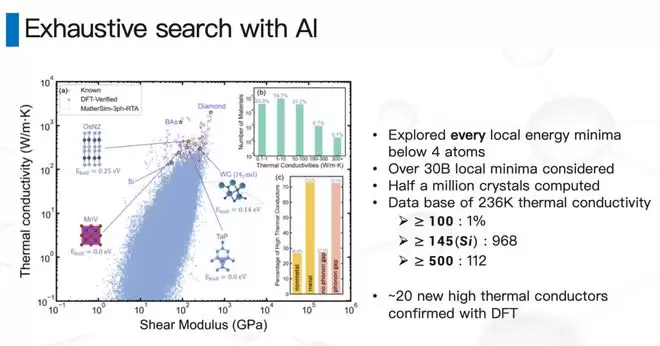
结论三:刚刚说了,我们做这件事的过程中其实做了一个暴力穷举——用数学的方法叫筛法。所以原则上,在一个给定的置信概率下,金刚石就是最高的。金刚石就在最上面——我们把每一个材料都看了一遍,金刚石就是我们数据库里最高的。
用一个比较技术性的说法:在常温常压下、无机晶体、体相材料里,金刚石基本上就设定了热导率的上限。做了一段时间之后,我们把前面那个前缀去掉了——金刚石基本上就是最高的,大家不用再花时间去找了。这对我们来说,是过去几年觉得做得比较有意义的一个结论。
反向生成材料
刚刚讲的是正向模型——它能帮我们看得更快,甚至快到把所有东西看一遍。但有些问题,我们不可能把全空间看完。人类已知的材料大约有20万种,但真正的材料空间高达10¹²量级——模型再快,也不可能全空间扫完。
那能不能更快地找到目标材料?我们用的方式是生成式AI。
团队最早做了一个叫Distributional Graphormer的工作。最初在碳材料和催化剂上做了一个概念验证,结果发现:在图学习加扩散模型的框架下,原则上可以做到“指哪儿打哪儿”——给定一个目标性质,模型就能生成出具有该性质的小分子、小分子晶体或晶体。一旦证实这条路可行,对机器学习来说,后面的事情就相对简单了:做大规模、投入算力——这就是MatterGen。我们把人类合成过的所有无机晶体材料(ICSD),加上数据库里能量较低的一些结构,拿来训练了一个可扩展的扩散模型。
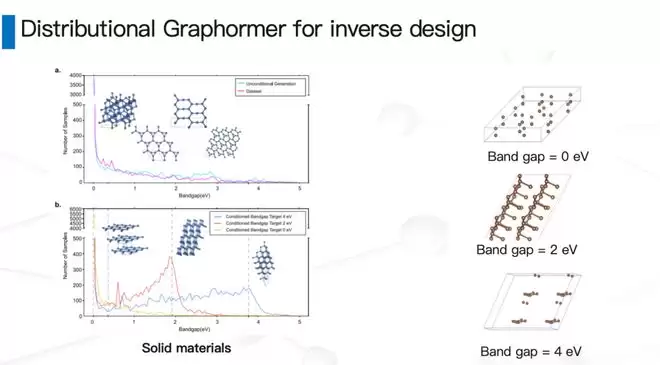
MatterGen有几个鲜明的特点。第一,它是一个在整个周期表上通用的生成模型。第二,它可以按照我们想要的性质进行条件约束生成。比如:
- 约束化学组分:如果你的实验室里只有碳、钴、氧、锂这几种元素,那就让模型只在这个空间里生成,它就有可能生成出钴酸锂——你不需要自己那么聪明。
- 约束对称性:比如要求一个四方晶系或者某种特定相,这在统计物理和凝聚态物理研究中比较有用。
- 约束标量性质:比如要一个不含稀土金属、单位体积下磁密度较高的材料——也就是我们常说的“少稀土金属、供应链稳定的强磁体”——同样可以设计。
不过,MatterGen也有它的局限。这类生成模型虽然有效,但它本质上是在高维隐空间里做插值,并没有真正做外推。这个结论我们目前还没有严格证明,但从长期实践来看确实如此。
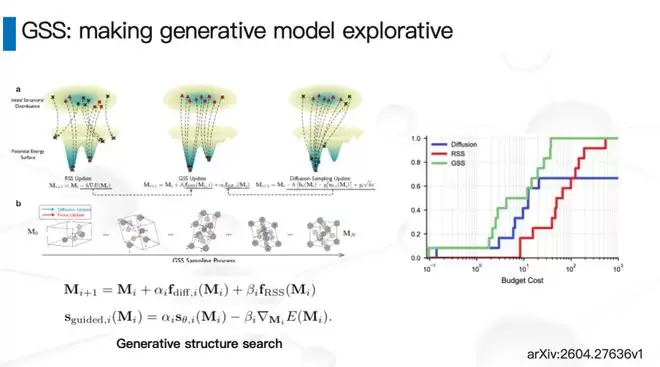
那么,有没有办法引入一些外部信号,让生成模型拥有更强的探索性?我们最近做了一个叫GSS(Generative Structure Search)的工作。通过算法改进,这个生成模型在理论上能够覆盖所有可能的材料。模型内部设计了一个可以调节的“旋钮”:把探索性调强一些,搜索会变慢,但能覆盖全空间;把利用性调强一些,生成速度更快,但新颖性会弱一些。有了这个调节机制,我们在很多案例上既获得了极高的搜索效率,又保证了足够的新颖性。
从理想晶体到工业材料
前面讲的MatterSim和MatterGen,处理的全是非常理想的情况——理想晶体、理想小分子,结构完美、成分纯净。这些在学术研究中很有价值,但距离真正能卖得出钱的工业材料,还有一段不小的距离。
为什么会有距离?至少存在两个问题。
第一个问题:前面那类纯模型全部是从零开始训练(train from scratch),人类历史上大量化学家的经验、文献里积累的知识,完全没有办法注入进去。
第二个问题:下游的工艺条件也很难被考虑进去。纯物理模型在这方面天然受限——它知道一个材料在理想状态下性质如何,但不知道在真实的合成环境、掺杂条件、加工工艺下会变成什么样。
那怎么把这些宝贵的先验知识结合进来?
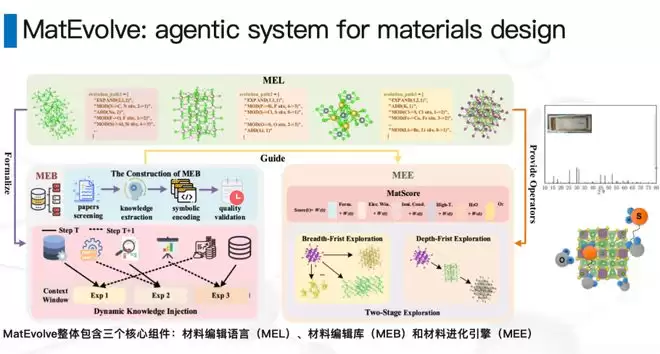
我们几位同事一起做了MatEvolve:它是一套智能体系统(agent system),具备两个核心能力:第一,可以参考文献;第二,可以把化学专家的专业知识结合进去。在这套系统的帮助下,我们用比较少的训练资源就能获得相当强的泛化能力。
做完这个模型之后,我们真的把它用在了下游场景中——用它设计了一个新材料,并且成功合成出来了。具体内容目前还没有完全披露,但这件事本身证明了一条路径:从AI设计到真实材料的闭环是可以走通的。
有了MatEvolve这样的智能体系统,我们实际上获得了一个环境:可以快速把人类历史上的知识注入到AI系统里。这里有一个很大的好处——我们不需要像做大语言模型那样,去做昂贵的中期训练或者强化学习。那些方法比较麻烦,成本也高。而有了智能体,我们可以用相对便宜、效果也不错的方式来做这件事,不用再没有任何先验地从零开始。相反,我们把过去这么多年积累的思路精华结合进来,把工艺条件也一并考虑进去。
这意味着,从理想晶体到工业材料,AI正在一步步填平中间的鸿沟。
最后,我们希望在未来两三年内,真的能做到这样一幕:我作为一名材料科学家,坐在办公室里,随口说一句:“Hey JARVIS,能不能帮我设计一种超级合金,用来制造下一代合金战甲?”
听起来像科幻电影吗?但这件事正在走向现实。
互动提问
Q1:你们得出的“常温下没有热导率超过金刚石的材料”的这个结论,它符合你的预期吗?你觉得可靠吗?
陆子恒:这是个挺有意思的事。我们做这个问题之前,发现凝聚态物理、统计物理、无机化学各个领域里,每个人——甚至非常资深的科学家——对这个问题的认识非常极端化。有的老师非常明确地告诉我们:“这玩意不存在,不用看,肯定不存在,不需要做。”但也有很多人直到今天仍然认为:“不,你不可能穷尽,一定还有新的。”
对我们来说,我们做到的是:在我们指定的空间内——虽然这是一个非常大的空间,也是最有可能存在的空间——以最高的执行概率把它穷尽了,并给出了一个相对较高的置信概率,告诉你在什么情况下这个东西不会存在。这是科学的说法。
但站在我个人角度,我们这个团队做了将近两年,我们的感受是:在常温常压、块体材料里,不太可能突破这个点了。
Q2:你们的模型擅长生成完美晶体,但大部分有用的晶体都是有缺陷或掺杂的,这个问题怎么解决?
陆子恒:很多材料的性质,一大部分内在性质取决于纯净晶体本身;但还有相当一部分——甚至我们非常关心的一部分——恰恰取决于那个非常微量的掺杂。如果你做半导体或者合金领域,这个问题尤其突出。我们在做模型的过程中,其实可以把这种考量做进去。但在筛选材料时,通常分两步走:
第一步,先把母体的、纯净的晶体筛选出来;第二步,再去筛选它的掺杂和各种缺陷的可能性。这些都是能做的。
但是再往后走一层——涉及到宏观、介观的跨尺度建模——到今天为止,我认为要做泛化性强的模型难度还比较大,还没有看到特别好的路。
Q3:10年后的材料实验室会是什么样子?
陆子恒:生物在实验室和AI上的发展比材料早个三四年、四五年,整个市场的进度也是如此。材料实验室的发展阶段差不多,但整体速度可能会稍微快一点,因为生物已经打了一个样板。
不过材料也有自己的难点,材料不像医药——药物的整个管线非常相似,基本上都是从先导化合物到hit到PCC到临床一期、二期、三期。但材料呢,种类非常多。所以真正做通用的材料平台时,会遇到一些困难。
时间上不一定更快,但我对这件事非常有信心——因为市场非常大,市场会催促整个实验流程以最快的速度加速。
作者简介
陆子恒,开物纪创始人,北京中关村学院首席材料科学家。主要研究大规模深度学习与其在材料设计领域的应用。于2018年从香港科技大学获得博士学位。之后,在包括耶鲁大学、中国科学院、英国法拉第研究所和剑桥大学等从事研究工作,曾任微软研究院首席研究员、微软科学智能中心材料方向负责人。主要工作包括:材料大模型MatterSim,在MatBench、MatBench Discovery等材料设计任务上取得榜首;IDEAL主动学习算法,取得了百万量级原子的在线化学精度模拟;DiG、MatterGen等主流材料生成模型。




