单细胞RNA测序技术的广泛普及,使得WGCNA(加权基因共表达网络分析)从传统的bulk RNA-seq分析成功延伸至单细胞转录组领域。hdWGCNA正是为此场景量身定制的R包,其高度模块化的设计能够高效构建细胞层次或空间层次的共表达网络,精准识别高度共表达的基因模块,并借助统计检验与生物学知识资源赋予这些模块生物学意义。该包的输入格式为研究者熟悉的Seurat对象,使用非常便捷。此前我们已详细讲解过hdWGCNA的基础分析流程,本次更新不仅优化了整体流程,还补充了网络可视化的实用内容。示例数据来自一篇2026年发表于Nature Communications的文章(Bean, J.C. et al., Sex-specific differences in mediobasal hypothalamus in response to nutritional states, Nat Commun 17, 2941, 2026),该文章本身即采用了hdWGCNA方法,是学习其应用思路的绝佳案例。原始数据量较大,为便于演示,我们进行了分层抽样——对每种celltype随机抽取10%的细胞,确保抽样后每种celltype仍有充足的细胞数量用于后续分析。
library(Seurat)
setwd("/home/data_analysis")
GSE282955_ARH_Sex_by_Nutr <- readRDS("~/data_analysis/GSE282955_ARH_Sex_by_Nutr.rds")
dim(GSE282955_ARH_Sex_by_Nutr)
#[1] 35318 93282
# 进行分层抽样,按细胞类型保留10%的细胞
set.seed(123)
cells_keep <- GSE282955_ARH_Sex_by_Nutr@meta.data %>%
rownames_to_column("cell") %>%
group_by(cell_type) %>%
sample_frac(size = 0.1) %>%
ungroup() %>%
pull("cell")
GSE282955_ARH_Sex_by_Nutr_sub <- subset(GSE282955_ARH_Sex_by_Nutr, cells = cells_keep)
DimPlot(GSE282955_ARH_Sex_by_Nutr_sub, label = T) + NoLegend()
dim(GSE282955_ARH_Sex_by_Nutr_sub)
#[1] 35318 9330
sa ve(GSE282955_ARH_Sex_by_Nutr_sub, file = 'GSE282955_ARH_Sex_by_Nutr_sub.RData')
2 安装包
hdWGCNA包依赖较多,推荐直接从GitHub安装,安装过程中几乎需要更新所有依赖包。WGCNA涉及不少专业术语,在此先简要回顾,以便更好地理解分析结果。此前我们在bulk RNA的WGCNA教程中已有详细注释,本部分仅作快速梳理。
# devtools::install_github('smorabit/hdWGCNA', ref='dev')
library(hdWGCNA)
package.version('hdWGCNA')
| 术语(Term) | 定义(Definition) |
|---|---|
| 共表达网络(Co-expression network) | 共表达网络定义为无向加权基因网络。节点对应基因表达谱,边由基因表达之间的成对相关性决定。通过将相关系数绝对值提升至幂次β≥1(软阈值化),加权基因共表达网络可突出高相关性而弱化低相关性。 |
| 模块(Module) | 模块是高度互连基因的聚类。在unsigned网络中,模块对应高绝对相关性的基因聚类;在signed网络中,则对应正相关的基因。 |
| 连接度(Connectivity) | 每个基因的连接度定义为它与网络中其他基因连接强度的总和,衡量该基因与所有其他基因的相关程度。 |
| 模块内连接度(Intramodular connectivity kIM) | 衡量给定基因与特定模块内其他基因的连接或共表达水平,可解释为模块成员资格的度量指标。 |
| 模块特征基因(Module eigengene E) | 模块特征基因是模块的第一主成分,代表该模块中基因的表达谱。 |
| 特征基因显著性(Eigengene significance) | 将模块特征基因与性状变量做相关性分析,所得相关系数即称为特征基因显著性。 |
| 模块隶属度(Module Membership, kME) | 对于每个基因,将其表达谱与模块特征基因进行相关性分析,得到“模糊”的模块隶属度量。例如MMblue(i)=cor(xi, Eblue),值越接近±1,表示该基因与蓝色模块高度连接;值接近0则不属于该模块。MM与kIM高度相关。 |
| 枢纽基因(Hub gene) | 共表达模块中高度连接的基因(连接度高的基因)。 |
| 基因显著性(Gene significance GS) | 将外部信息纳入网络,用基因显著性度量。|GS|越大,该基因的生物学意义越重要。 |
| 模块显著性(Module significance) | 模块中所有基因的|GS|平均绝对值。当GS定义为基因与性状的相关时,模块显著性与模块特征基因-性状相关性高度一致。 |
3 数据分析
3.1 分析准备
首先加载必要的R包和数据。
library(hdWGCNA)
library(WGCNA)
library(Seurat)
library(tidyverse)
library(igraph)
library(cowplot)
library(patchwork)
library(dplyr)
library(ggplot2)
library(stringr)
setwd('/Users/ks_ts/Documents/公众号文章/hdWGCNA复现/')
load("./GSE282955_ARH_Sex_by_Nutr_sub.RData")
# 保存原始数据对象
seurat_obj <- GSE282955_ARH_Sex_by_Nutr_sub
hdWGCNA的输入必须是Seurat对象,请注意将其更新到最新版本,以避免Seurat v4/v5的兼容性问题。
seurat_obj <- SetupForWGCNA(
seurat_obj,
gene_select = "fraction", # 使用所有基因,但要求基因至少在5%的细胞中表达
fraction = 0.05,
wgcna_name = "ARH" # 本次分析的项目名称
)
接下来构建metacells,这一步旨在降低单细胞数据的噪声,提升共表达分析的稳健性。
seurat_obj <- MetacellsByGroups(
seurat_obj = seurat_obj,
group.by = c("cell_type3", "Batch"),
reduction = 'pca',
k = 25,
ident.group = 'cell_type3'
)
seurat_obj <- NormalizeMetacells(seurat_obj)
3.2 共表达网络分析
在NC文章中,作者重点关注Agrp细胞中的基因表达模块,因此构建网络时仅选择这一种细胞类型。通过group_name参数指定目标细胞类型,后续分析将基于Agrp的表达矩阵进行。
seurat_obj <- SetDatExpr(
seurat_obj,
group_name = "Agrp",
group.by = 'cell_type3',
assay = 'SCT',
layer = 'data'
)
接下来是WGCNA分析中最关键的一步——选择软阈值。
seurat_obj <- TestSoftPowers(
seurat_obj,
networkType = 'signed' # 生物学数据更适合signed网络,以明确区分正负调控关系
)
plot_list <- PlotSoftPowers(seurat_obj)
wrap_plots(plot_list, ncol=2)
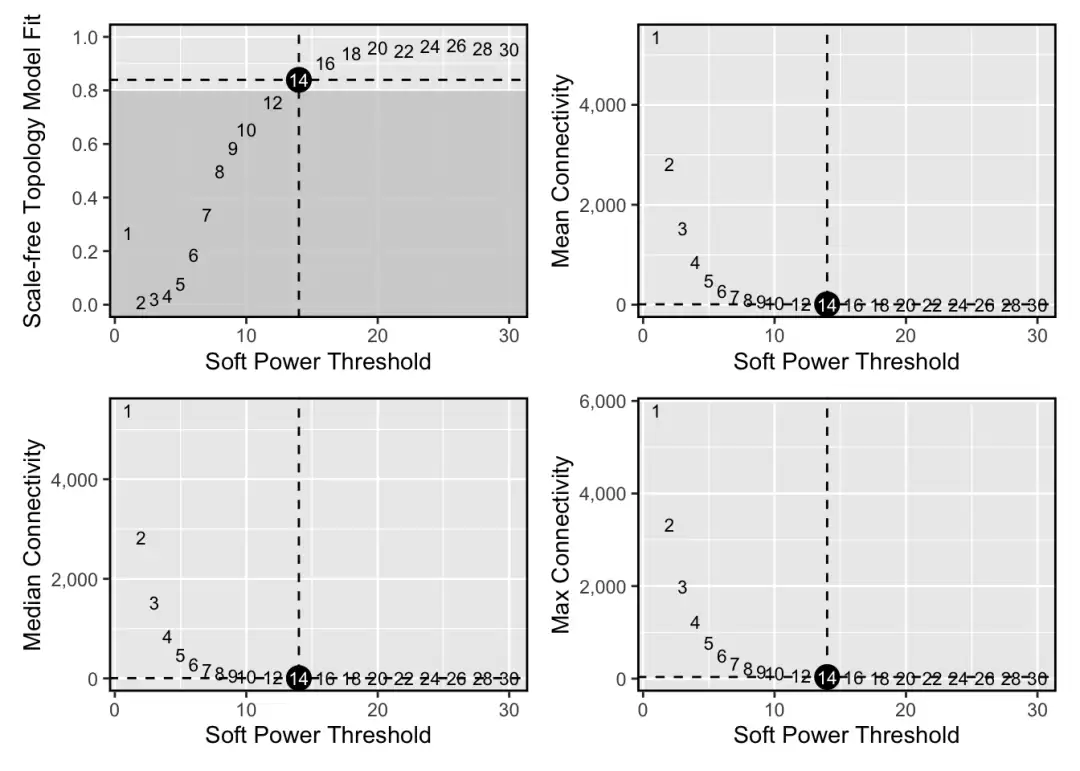
选定软阈值后,使用ConstructNetwork函数构建共表达网络。该函数底层实际上调用了WGCNA的blockwiseConsensusModules,相关参数可以直接传递。
seurat_obj <- ConstructNetwork(
seurat_obj,
soft_power = 15,
setDatExpr = FALSE,
tom_name = "Agrp'
)
PlotDendrogram(seurat_obj, main='Agrp hdWGCNA Dendrogram')

3.3 模块特征基因与连通性
模块特征基因(Module Eigengenes, MEs)用于概括整个模块的基因表达谱。
seurat_obj <- ScaleData(seurat_obj, features = VariableFeatures(seurat_obj))
seurat_obj <- ModuleEigengenes(seurat_obj, group.by.vars = "Batch")
hMEs <- GetMEs(seurat_obj)
然后计算每个基因基于特征基因的连通性(kME),用于后续筛选hub基因。
seurat_obj <- ModuleConnectivity(
seurat_obj,
group.by = 'cell_type3',
group_name = 'Agrp'
)
seurat_obj <- ResetModuleNames(
seurat_obj,
new_name = "Agrp_M" # 原始模块名称是数字,加上前缀使其更清晰,例如 Agrp_M1, Agrp_M2 ...
)
modules <- GetModules(seurat_obj) %>% subset(module != 'grey')
3.4 重新设置模块颜色
默认情况下每个模块分配一种颜色,但为了排版统一,我们可以自定义颜色方案。
modules <- GetModules(seurat_obj)
mod_colors <- dplyr::select(modules, c(module, color)) %>%
distinct %>%
arrange(module) %>%
.$color
mod_colors
# 共有14个模块,分配一组自定义颜色
my_color = c("#D52126", "#88CCEE", "#FEE52C", "#117733", "#CC61B0",
"#99C945", "#2F8AC4", "#332288", "#E68316", "#661101",
"#F97B72", "#DDCC77", "#11A579", "#89288F")
seurat_obj <- ResetModuleColors(seurat_obj, my_color)
4 可视化
4.1 基础可视化
重新设置颜色后,可以重新绘制聚类树,以便更直观地展示模块层次结构。
PlotDendrogram(seurat_obj, main='Agrp hdWGCNA Dendrogram')

按照kME排序,可视化每个模块的hub基因分布。
PlotKMEs(seurat_obj, ncol = 4)
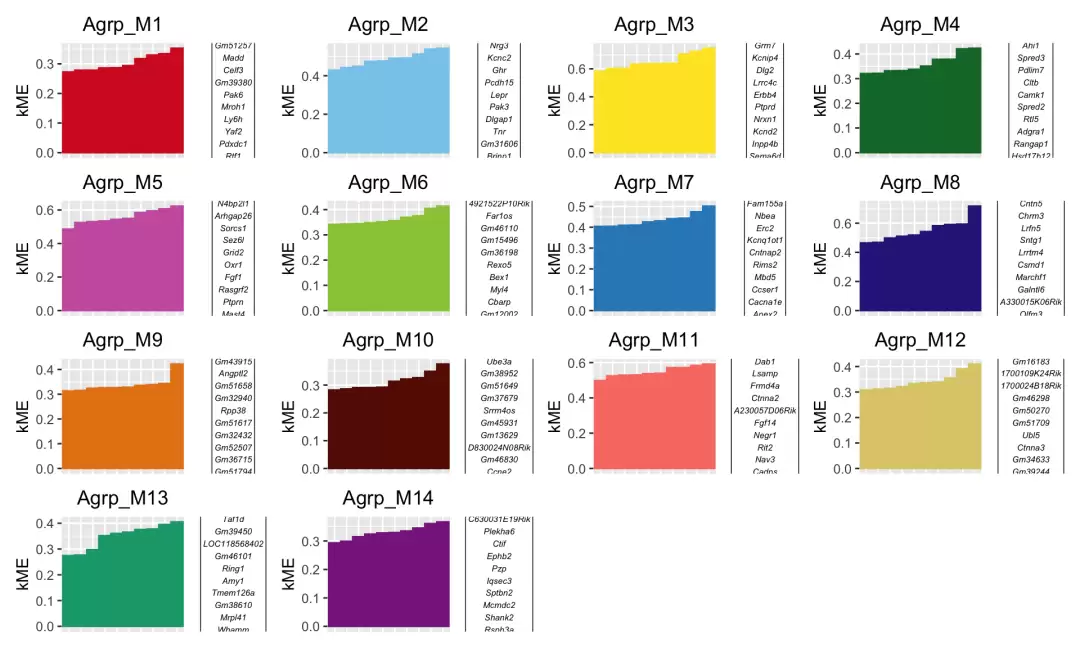
计算模块基因集评分(选取kME排名前25的hub基因),并使用UCell方法进行评分。
library(UCell)
seurat_obj <- ModuleExprScore(
seurat_obj,
n_genes = 25,
method = "UCell'
)
plot_list <- ModuleFeaturePlot(
seurat_obj,
features = 'scores',
order = 'shuffle',
reduction = 'tsne',
ucell = TRUE,
point_size = 0.1
)
wrap_plots(plot_list, ncol = 4) + plot_annotation(title = "Hub genes signature scores")

4.2 网络可视化
hdWGCNA提供了多种网络可视化方式。ModuleNetworkPlot可为每个模块单独绘制网络(默认显示前25个基因),HubGeneNetworkPlot则绘制包含所有模块的综合网络(每个模块按指定数目的hub基因展示),而ModuleUMAPPlot利用UMAP降维展示所有基因的共表达网络。
# 将所有模块的网络导出至指定文件夹
ModuleNetworkPlot(seurat_obj, outdir = "./Agrp_all_ModuleNetworks')
# 单独绘制某个模块,例如Agrp_M4
ModuleNetworkPlot(
seurat_obj,
mods = 'Agrp_M4',
n_inner = 10,
n_outer = 15,
n_conns = Inf,
vertex.label.cex = 1
)
HubGeneNetworkPlot(
seurat_obj,
n_hubs = 10,
n_other = 5,
edge_prop = 0.75,
mods = c('Agrp_M1', 'Agrp_M2', 'Agrp_M4', 'Agrp_M6', 'Agrp_M7', 'Agrp_M8', 'Agrp_M9')
)
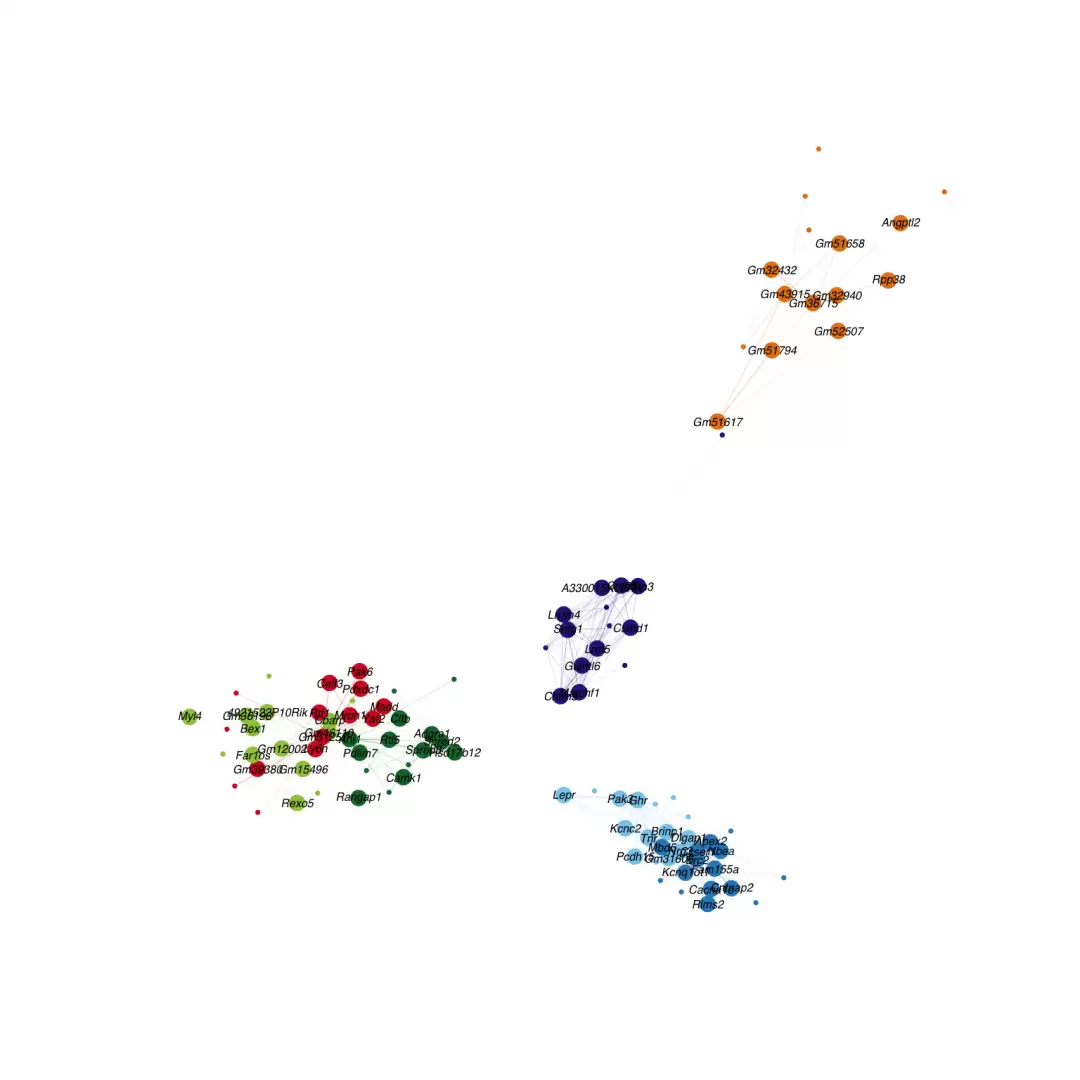
seurat_obj <- RunModuleUMAP(
seurat_obj,
n_hubs = 10,
n_neighbors = 15,
min_dist = 0.3
)
ModuleUMAPPlot(
seurat_obj,
edge.alpha = 0.5,
sample_edges = TRUE,
edge_prop = 0.1,
label_hubs = 3,
keep_grey_edges = FALSE
)

上图与原文效果存在一定差距,因此我们编写了一个自定义函数,用于复现NC文章Supplementary Fig. 19的所有元素。核心要点包括:采用UMAP展示网络,每个网络显示top hub基因,不同模块使用不同颜色;用框线圈定模块范围并添加背景底色;自动添加模块名称,文字颜色与模块颜色保持一致。当然,该函数仍有诸多细节可以进一步优化。

ks_hdWGCNA_UMAPnet(
seurat_obj = seurat_obj,
wgcna_name = "ARH',
edge_prop = 0.05,
top_hub = 3,
r_text = 10
)





