
一篇关于全原子扩散模型 AGDIFF、非天然氨基酸环肽与药物设计瓶颈的解读
想象一个药物分子,它就像一枚小巧精致的“手环”,由几个氨基酸首尾相连,围成一个结构受限的环。这种环状结构,让它比普通线性肽更稳定,也更容易在复杂的生物环境中保持自己的形状。如果再给它换上一些“非天然”的氨基酸,它甚至能获得更好的亲和力、代谢稳定性和跨膜能力。问题在于,这类分子越是接近理想中的药物,它的结构就越不像教科书里那些标准的蛋白质。它们小巧、刚性、改造多,常常带着 D-氨基酸、N-甲基化残基、内酯键、β-氨基酸和各种不常见的侧链。对于传统的蛋白质结构预测模型来说,这带来了一个非常现实的尴尬:它们熟悉的“序列语言”,在这里行不通了。
今年4月,上海交通大学药学院和张江高等研究院的团队在《Journal of Chemical Information and Modeling》上发表了一篇论文。论文的核心,是将一个原本用于小分子构象生成的全原子扩散模型 AGDIFF,重新训练到了大环肽构象数据集 CREMP 上。这样一来,模型就能直接从二维分子图出发,生成那些含有非天然氨基酸的小环肽的三维构象集合,并通过立体化学校正,来解决生成模型中常见的手性错误问题。
论文给出的核心结果非常直接:在 CREMP 内部测试集上,AGDIFF 的平均最小 RMSD(均方根偏差)达到了 0.79 Å,环扭转指纹偏差 rTFD 为 6.55°;而在更具挑战性的外部非天然氨基酸泛化测试集中,RMSD 为 1.04 Å,rTFD 则升至 15.27°。这个结果清晰地表明,模型已经能够较好地恢复小环肽的整体三维拓扑结构;但同时也提示我们,对于那些训练集中没有覆盖到的内酯键、β-氨基酸等化学模式,模型仍然会付出局部扭转角误差显著升高的代价。
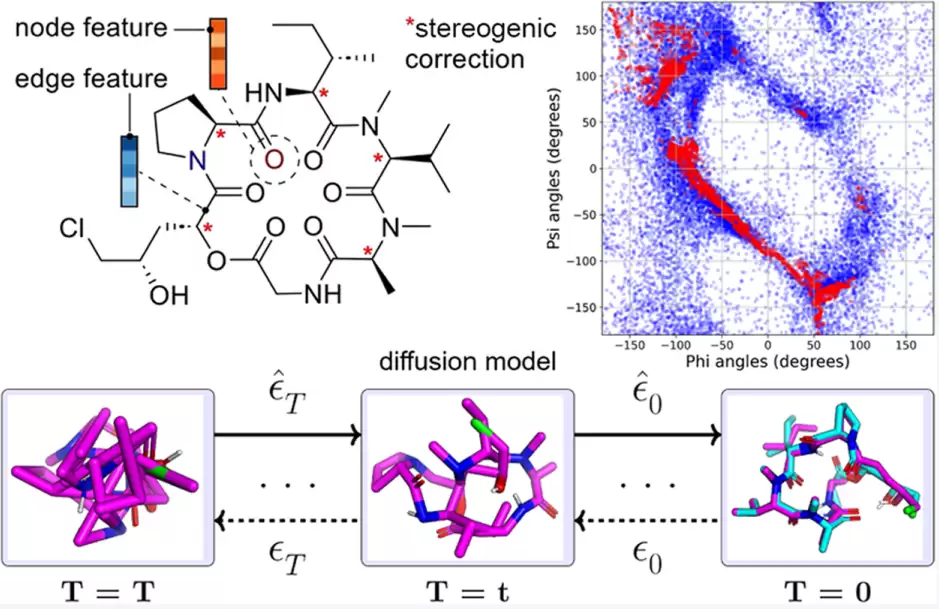
AGDIFF 从二维分子图出发,通过扩散去噪生成含非天然氨基酸小环肽的三维构象,并结合立体化学校正提高生成结构的手性一致性。
为什么小环肽正在变得重要?
在肽类药物这个大家庭里,环肽是一个非常特殊的分支。线性肽就像一根柔软的链条,容易被蛋白酶切割,在水溶液里也摇摆不定;而环肽则把链条的头尾扣在了一起,形成了一个受限的环状骨架。这种环化拓扑结构,极大地压低了构象的自由度,让分子更容易维持一个稳定的三维形状。这对药物发现来说,是个非常关键的特性:药物并不是靠序列去识别靶点,而是靠空间中的氢键供体、氢键受体、疏水斑块、电荷分布和形状互补来精准结合靶点。
环肽的另一个吸引力,在于它恰好位于传统小分子和抗体之间的“中间地带”。小分子通常擅长钻进深口袋,但面对蛋白-蛋白相互作用这类大而平的界面时,常常会因为没有足够的接触面积而束手无策。抗体虽然结合面积大,却很难进入细胞内部。环肽的体量和形状,给了它一种折中的可能性:它可以展开一片较大的结合表面,又有机会通过结构预组织、N-甲基化、D-氨基酸替换、侧链改造等方式,来提高自身的稳定性和跨膜潜力。
根据文章的说法,截至 2024 年,已经有 42 个环肽药物获得了 FDA 批准,约占所有已上市肽类药物的近一半;其中 33 个含有非天然氨基酸。作者举出的例子包括用于 T 细胞淋巴瘤治疗的 Romidepsin、抗菌药 Vancomycin、免疫抑制剂 Cyclosporine A,以及用于斑块状银屑病的 Icotrokinra。在临床管线中,Enlicitide 和 Paluratide 也被作为小环肽候选物提及。
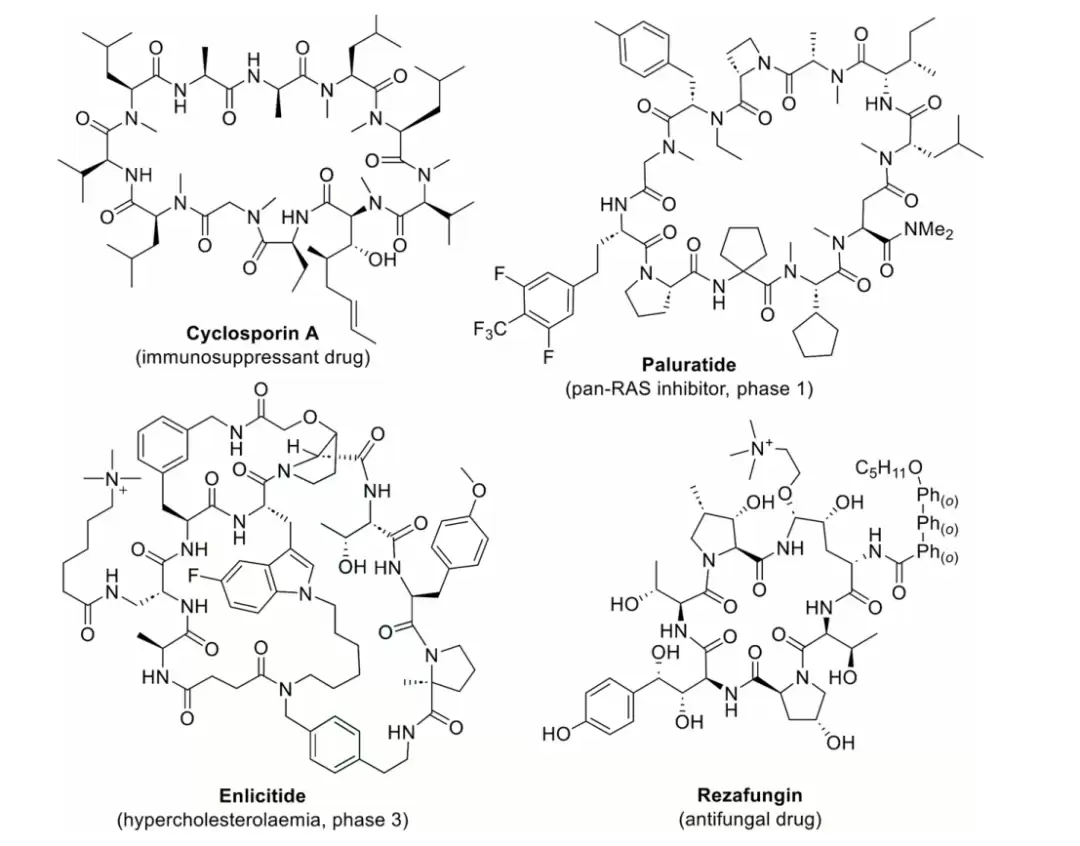
代表性含非天然氨基酸的大环肽药物及临床候选分子,包括 Cyclosporine A、Paluratide、Enlicitide 与 Rezafungin。
药物化学家真正关心的环肽,往往不是那些由标准氨基酸排成的漂亮小圆圈。为了让分子更像药,研究人员会不断对它进行改造:引入 D-氨基酸来抵抗酶解,用 N-甲基化来减少氢键暴露,换上疏水侧链增强膜相容性,甚至用非肽键的连接方式改变环的力学性质。很多小环肽还具有所谓的“分子变色龙”行为:在水相中,它们会暴露极性基团以保持溶解性;而在脂质环境中,它们又通过构象切换,把极性主链埋藏起来,从而提高被动跨膜渗透的机会。论文特别提到,常被讨论的被动跨膜环肽代表 Cyclosporine A 由 11 个残基组成,这也提示了小尺寸和构象集合对口服或细胞内递送来说是多么重要。
计算问题就卡在这里了。对于大蛋白而言,AlphaFold 系列、RFdiffusion、ProteinMPNN 等模型已经彻底改变了结构预测和设计的方式;在环肽方向,Baker 等团队也通过循环偏移等策略推动了深度学习设计。但这些模型的基础知识,大部分都来源于蛋白质结构数据库。它们更熟悉那些体型更大、二级结构更清晰、更像蛋白质的环肽结合物。而面对小环肽,尤其是那些残基数小于 10、又大量包含非天然骨架和侧链的分子时,序列模型会遇到两层困难:一是数据稀缺,二是化学表达不够精细。
论文中,作者系统挖掘了 PDB 数据库后发现,具有实验结构的小环肽唯一序列只有 344 条。对于一个需要学习立体化学、环张力、顺反肽键、非天然残基和多构象平衡的模型来说,这样的数据量显然是远远不够的。于是,一个更自然的思路便出现了:既然药物化学家手里首先有的是二维结构式,那就从化学图开始,而不是把小环肽硬塞进线性的氨基酸序列里。
从序列折叠转向化学图生成
AGDIFF 的基本思路其实非常直观:先把一个真实的三维结构逐步加噪,让它变成一团乱糟糟的坐标;然后,模型学习这个过程的逆向操作,从噪声中一步步恢复出合理的三维构象。这和图像扩散模型从噪声中生成图片的逻辑类似,只不过这里生成的是原子的坐标。
这篇论文保留了 AGDIFF 原有的主体架构,并将其重新训练到了环肽任务上。模型的输入不再是氨基酸序列,而是二维分子图:节点是原子,边是化学键。图中天然地包含了原子类型、连接关系、局部化学环境等信息。这样做有一个很实用的好处:环肽的序列表示存在循环排列歧义,同一个环从不同的残基开始读,会得到不同的线性序列;而二维分子图则完全没有这个问题。更重要的是,图表示天然就能容纳非天然氨基酸、侧链环化、骨架异构、杂原子连接等复杂的化学情况。
模型主要由两个部分组成。一个是分子特征提取模块,它用图神经网络来读取原子的连接和局部化学环境;另一个是扩散生成模块,它负责从噪声坐标中恢复几何结构。从论文的示意图来看,局部的 GIN 编码器用于捕捉键级相互作用,而注意力增强的 SchNet 编码器则用于捕捉长程的几何信息,两者共同给出构象梯度,指导反向扩散过程。
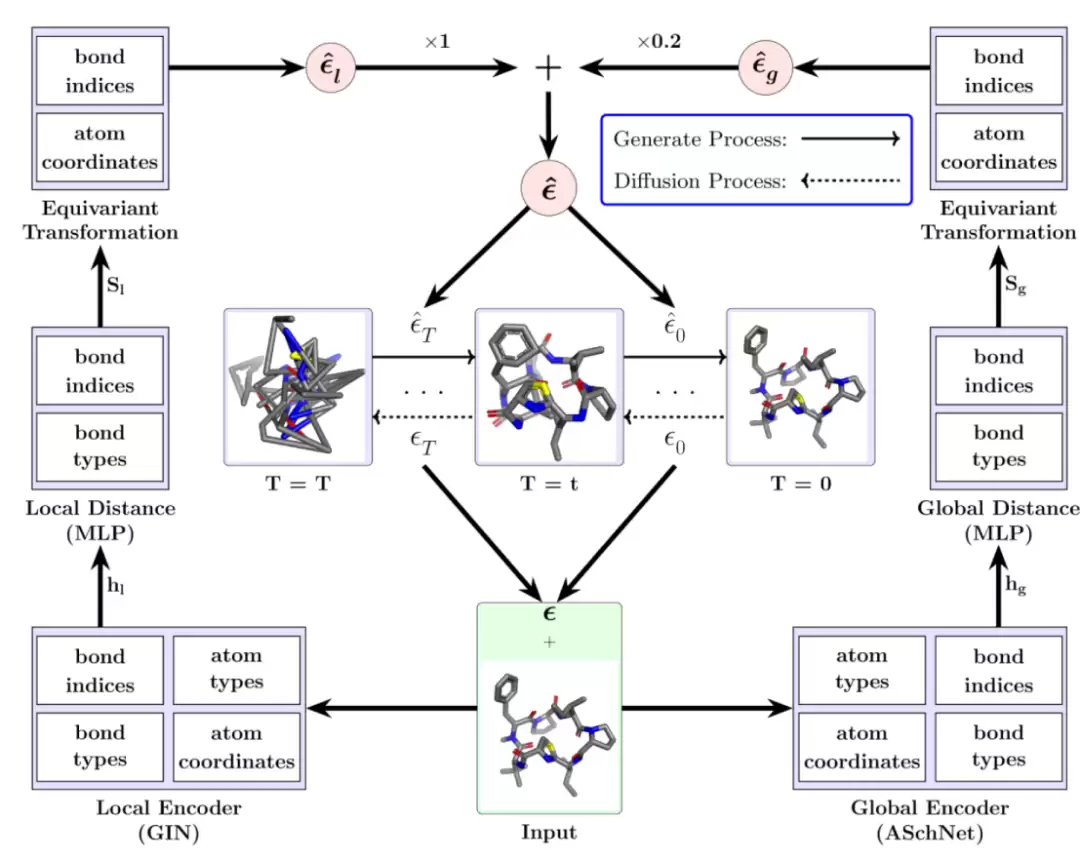
AGDIFF 使用局部图同构网络和全局注意力增强 SchNet 同时编码键级相互作用与长程几何信息,并通过扩散去噪生成小环肽三维构象。
在数据层面,作者使用的是 CREMP 数据集,全称是 Conformer-Rotamer Ensembles of Macrocyclic Peptides。这个数据集包含了 36,198 个唯一的大环肽,覆盖了天然氨基酸、D-氨基酸和 N-甲基化氨基酸。数据生成的流程结合了 RDKit 构象采样、GFN2-xTB 半经验优化和 CREST 构象集合精修,并通过与 NMR 衍生结构的 Ramachandran 分布比较,验证了其合理性。
训练和评估的拆分也相当清晰:28,800 个分子用于训练,3,600 个用于验证,200 个用于内部测试。此外,作者还从 CREMP-CycPeptMPDB 中挑选了 40 个更具非天然氨基酸挑战性的环肽,作为外部泛化测试集。这 40 个分子被特意选为可旋转键较少的分子,目的是减少过度柔性带来的干扰,把问题更集中地落在新化学模式的空间泛化上。
小环肽首先是化学对象,其次才是序列对象
这项工作的第一个关键转向,就是把小环肽放回到全原子化学图的语境中。对于标准蛋白,序列是一种非常高效的表达方式;但对于含有非天然氨基酸的小环肽,序列就显得有些笨拙了。一个 D-氨基酸、一个 N-甲基化残基、一个侧链到主链的连接、一个内酯键,都需要额外的字典或特殊标记才能表示。而化学图则直接表达了原子和键,让模型看到的是分子本身。
第二个转向,是强调构象集合。小环肽既不像刚性小分子,也不像大蛋白那样通常追求一个主导的折叠状态。很多环肽的药效、渗透性和选择性,恰恰源自一组可以互相转换的低能构象。论文在评估时,并没有简单地比较单个预测结构,而是对参考集合中的每个构象,在生成集合中寻找最接近的构象,再计算平均最小 RMSD 和 rTFD。这种评价方式,更贴近环肽真实的物理图景。
第三个转向,是把立体化学当成一个“硬”问题来处理。很多全原子生成模型在生成三维坐标时,会出现手性中心翻转的问题,尤其在没有显式立体约束的情况下,模型很可能生成镜像构象。对药物分子来说,这可不是小误差。一个手性中心反了,可能就意味着一个不同的分子,其药效、毒性和代谢都会随之改变。论文采用了一个轻量级的后处理校正方法:利用 RDKit 给出的立体化学标签作为参考,计算手性中心邻近原子的有向体积符号;如果符号不匹配,就通过几何反射来修正构型。如果结构中间出现超过一个手性不匹配的构象,则会被排除,以保证集合的可靠性。
第四个转向,是使用环肽专门的数据,而不是用泛化的小分子数据硬训。论文后面的消融实验结果非常有说服力:用 GEOM-Drugs 训练的 AGDIFF 虽然还能大致抓住整体拓扑,却无法稳定保持肽键 ω 角的顺式或反式平面几何;而用 QM9 训练时,模型会把大环肽压成不合理的球状结构。小分子数据集虽然体量很大,但它没有包含足够的大环肽几何先验。这里的经验非常清楚:数据的分布,决定了模型是否懂得某类化学语言。
实验结果:0.79 Å 的精度背后,真正要看三件事
论文使用了两个指标来评价结构质量。RMSD 衡量的是整体原子坐标的偏差,它反映了全局结构是否对齐;而 rTFD 衡量的是环骨架扭转角的偏差,它更关注环本身的构象是否正确。对于小环肽来说,两者都很重要:如果 RMSD 很好看但扭转角错了,可能意味着主链的姿态不对;反过来说,如果 rTFD 合理但侧链乱摆,也会影响对结合特征的呈现。
在 CREMP 内部测试集上,AGDIFF 达到了 0.79 Å 的平均最小 RMSD 和 6.55° 的平均最小 rTFD。对于环肽构象集合生成来说,这个量级已经相当可观。进一步按顺式肽键数量分组后,模型在含有 0 到 4 个顺式肽键的分子上,表现都相当稳定:无顺式肽键时,RMSD 为 0.77 Å、rTFD 为 6.34°;含 1 个顺式肽键时,RMSD 仍为 0.77 Å、rTFD 为 8.42°;即使在样本数很少的 3 个或 4 个顺式肽键组,RMSD 也没有出现明显的失控。
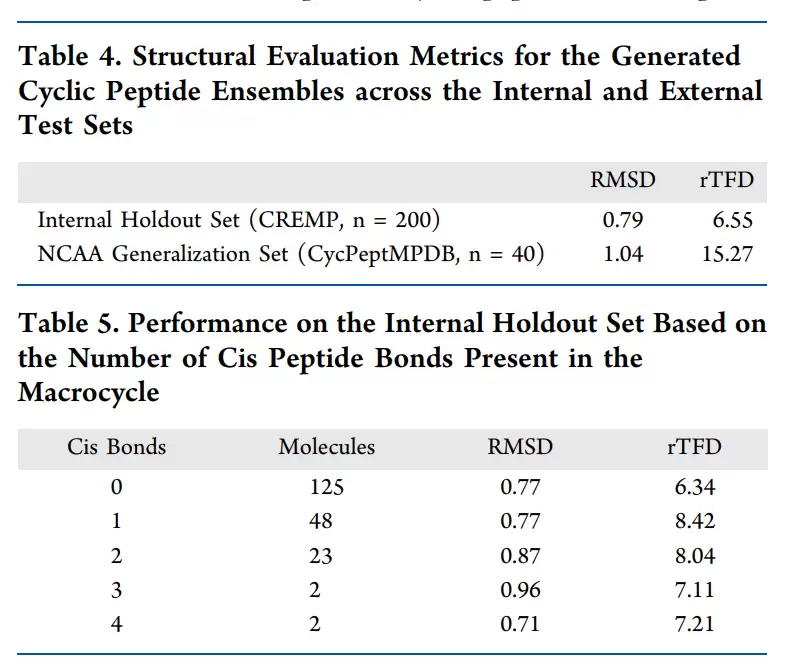
AGDIFF 在内部 CREMP 测试集和外部非天然氨基酸泛化测试集上的结构预测精度,以及不同顺式肽键数量下的性能表现。
外部泛化测试更能揭示模型的边界。CREMP-CycPeptMPDB 的 40 个环肽包含了更复杂的非天然氨基酸和环化化学。AGDIFF 在这里的平均最小 RMSD 升至 1.04 Å,整体拓扑仍然保持得较好;但 rTFD 却升至 15.27°。作者分析认为,主要原因在于外部测试集中间出现了训练数据没有覆盖的内酯键和 β-氨基酸。这表明,模型对新化学空间已经具备了一定的外推能力,但对于从未见过的连接方式,环扭转角的预测仍需付出代价。
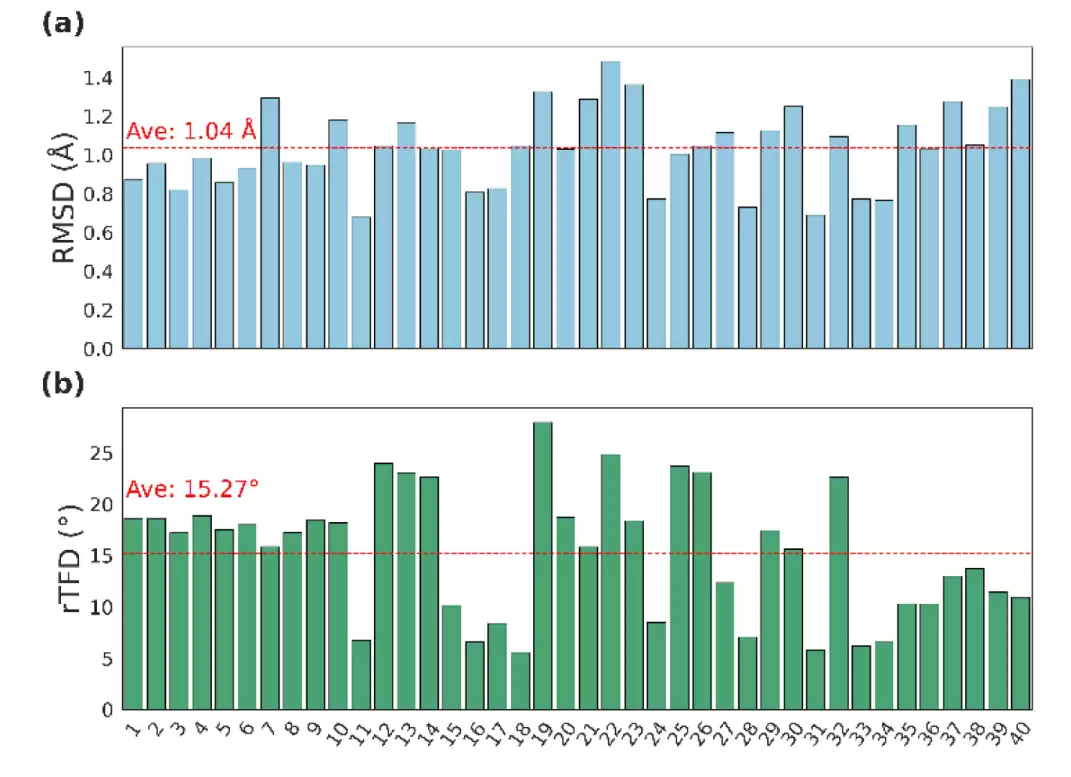
外部非天然氨基酸泛化测试集中,AGDIFF 的 RMSD 保持在约 1 Å 水平,但 rTFD 对训练集中缺失的内酯键和 β-氨基酸更敏感
论文还展示了 4 个外部测试分子的可视化叠合图。即使 rTFD 在复杂分子上有所升高,但生成的结构和参考的低能构象,依然能对齐出相似的整体环形拓扑。作者举到的例子,包括含有噻唑啉环的分子、含有 β-氨基酸并通过内酯键连接到氯取代非天然氨基酸的分子、主链里有 3 个内酯连接的分子,以及同时包含苯甘氨酸和 γ-吡咯烷基谷氨酸片段的分子。这个图给人的直观感受是:模型没有被复杂的化学结构完全打乱阵脚,但局部的扭转几何,还需要更丰富的数据来进一步校准。
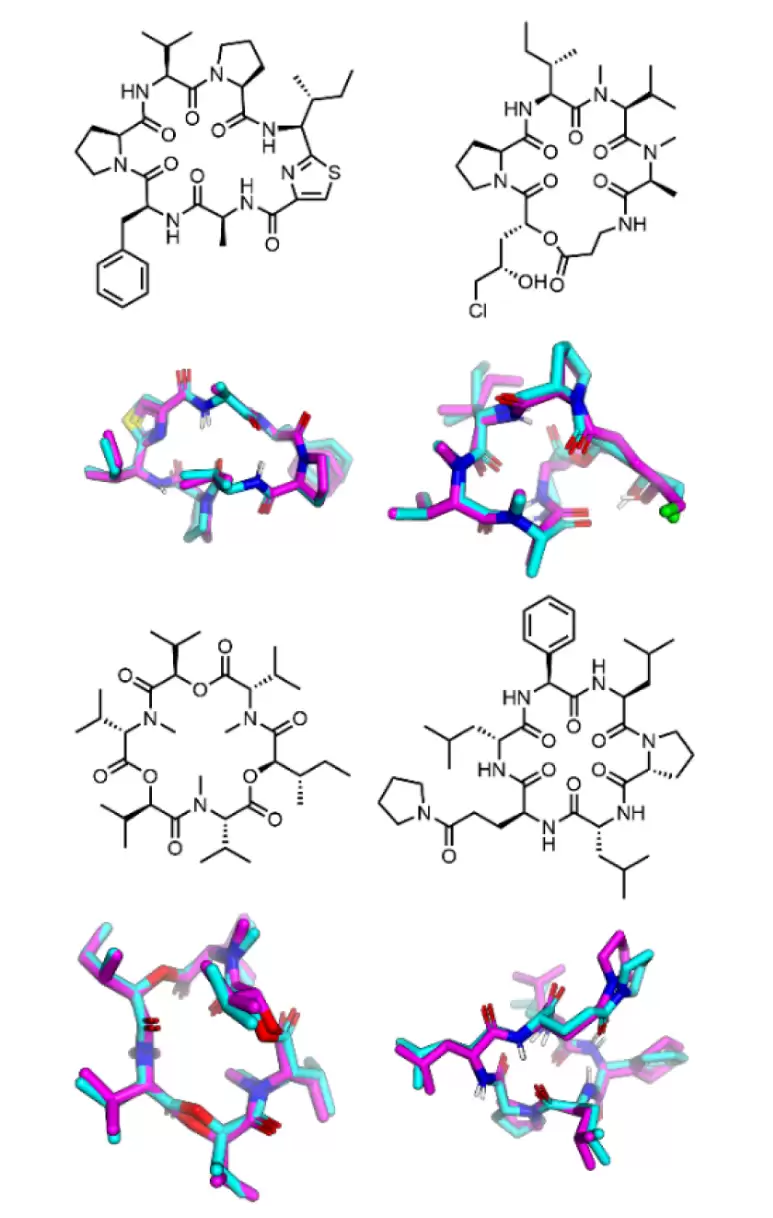
外部测试集中 4 个代表性非天然氨基酸环肽的生成构象与参考低能构象叠合,显示 AGDIFF 能较好恢复整体分子拓扑。
Ramachandran 图则从另一个角度检查生成的结构是否像真实的肽骨架。经校正后的模型,其生成的构象大多落在了合理的主链二面角区域,只有少量离群点。可以这样理解:Ramachandran 图就像一张主链运动的“地图”,如果大量点落在了不合物理直觉的区域,那说明模型可能只是把原子摆到了相似的位置,却并没有真正学会肽骨架的“构象语法”。
手性校正的效果则更加直接。论文显示,经过后处理立体化学校正,手性中心的正确率从 50.2% 提高到了 100%;与此同时,内部测试集的平均最小 RMSD 从 1.06 Å 降到了 0.79 Å,rTFD 从 9.10° 降到了 6.55°。这些数字清楚地表明,手性错误绝不是一个孤立的标签问题,它会真实地影响三维构象的整体对齐和主链扭转。
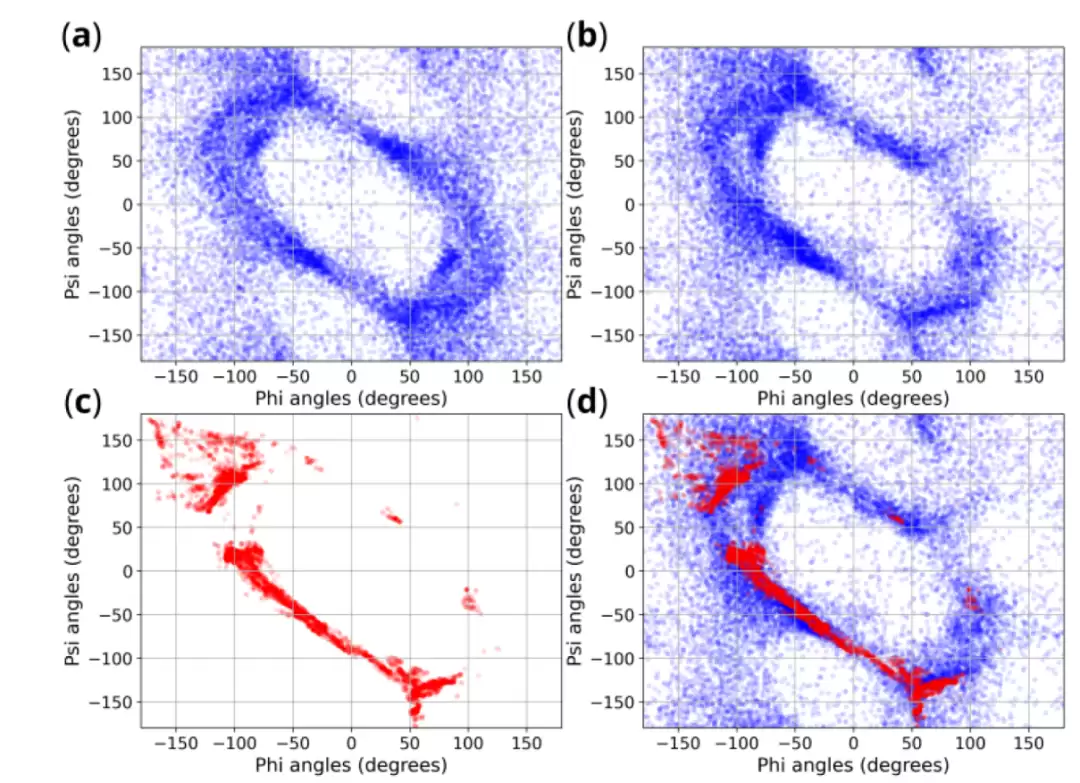
立体化学校正前后,模型生成构象的主链二面角分布与参考构象的相似性明显提高,说明手性校正有助于生成更合理的环肽构象集合。
论文还用一个实验结构 4B8Y 做了基准比较。这个分子被选中,是因为其中的非天然氨基酸能够被参与对比的主流模型残基字典所支持。作者最初考虑了 AlphaFold3、Boltz-2、Chai-1 和 Protenix;但在实际测试中,Chai-1 缺少生成环肽的能力,Boltz-2 虽然能处理输入,但生成的非天然氨基酸残基出现了物理上不合理的随机原子摆放。最终在可比较的结果中,AGDIFF 对 4B8Y 的最小 RMSD 为 1.98 Å,rTFD 为 18.5°;ETKDG3 为 2.32 Å 和 23.4°;AlphaFold3 为 2.80 Å 和 43.2°;Protenix 为 2.95 Å 和 57.5°。
这个结果不宜被解读为 AGDIFF 已经全面超越了所有蛋白质结构模型。它更像一个清晰的局部信号:在小尺寸、含有非天然氨基酸、需要全原子化学细节的环肽场景中,专门的化学图扩散模型,比以蛋白质为中心的模型要更合拍。尤其是当输入分子已经被药物化学家画成二维结构时,从图到构象集合的路径,就显得非常自然。
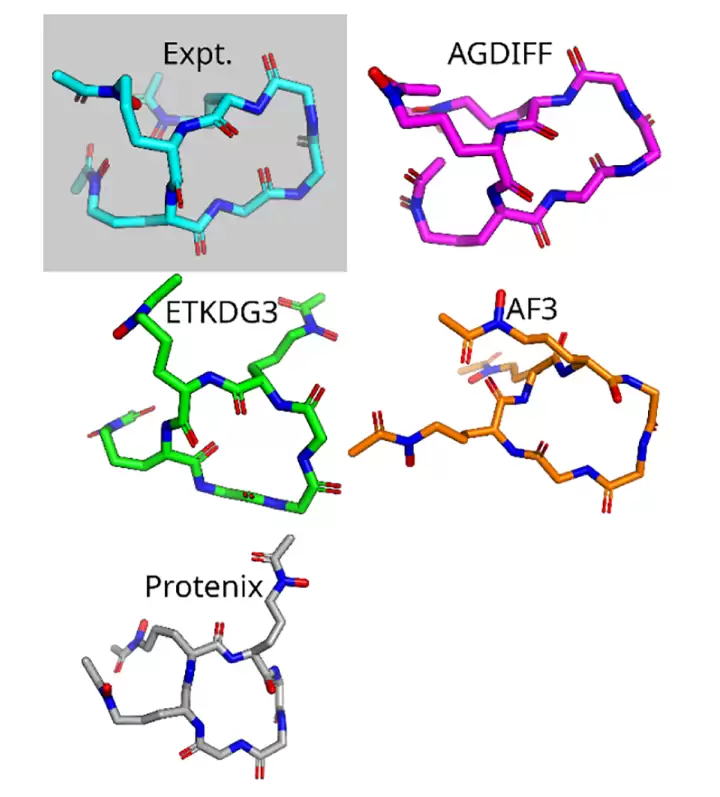
在含非天然氨基酸环肽 4B8Y 上,AGDIFF 相比 ETKDG3、AlphaFold3 和 Protenix 更接近实验参考结构。
启发
对于环肽药物设计来说,一个可靠的三维构象集合,可以成为许多下游任务的起点。药物化学家需要知道氢键供体和受体在哪里,疏水基团是如何排布的,主链有没有把极性原子暴露出来,某个侧链是否能伸到靶点的口袋里。AGDIFF 的价值正在这里:它能够快速地将二维分子图转化为三维构象集合,为分子对接、三维药效团建模、构象筛选和先导优化提供结构基础。
对蛋白-蛋白相互作用靶点来说,这一点尤其重要。许多 PPI 靶点缺少深口袋,传统小分子很难获得足够的接触面积;而环肽可以用它预组织好的环形骨架,呈现出一片更大的结合表面。如果模型能在早期就给出可信的构象,研究人员就能更快地判断某个环肽骨架是否具备正确的空间排列,从而减少盲目合成和反复试错。
另一个潜在方向是跨膜性设计。很多小环肽的成药性,不仅取决于它是否能结合靶点,还取决于它能否进入细胞、能否以合适的构象穿过脂膜。AGDIFF 本身并不直接预测膜渗透率,但它生成的构象集合,为识别“分子变色龙”行为提供了结构素材。一个分子在不同环境中是否能折叠起来遮蔽极性主链,是否能在水相和脂相之间切换姿态,最终仍需要结合物理模拟、实验测定和更专门的性质模型来分析。
这也是论文在未来的展望中提到的方向:把 AGDIFF 与图生成模型结合,做端到端的环肽设计;把立体化学约束直接写进训练目标,而不是只靠后处理;进一步从结构生成走向结构-功能预测,把构象灵活性、膜渗透性和靶点亲和力放到同一个设计框架里。
END:小环肽建模正在回到化学本身
这篇论文的价值,不只在于把 RMSD 做到了 0.79 Å。从更深层的角度看,它把那些含有非天然氨基酸的小环肽,从蛋白质序列的框架里解放了出来,转向了更贴合药物化学实际的二维分子图表示。对于小环肽来说,原子、键、手性、环张力、顺反肽键和构象集合,往往比线性序列更接近问题的本质。
AGDIFF 在 CREMP 数据集上学到的大环肽几何先验,让它有能力从噪声中恢复小环肽的三维形状;立体化学校正则确保了输出不再被镜像构型所拖累;外部测试集和 4B8Y 的对比结果则说明,这条技术路线对于复杂的非天然氨基酸分子,已经展现出了现实的潜力。它当然还不能替代实验,也无法直接回答活性和渗透性问题,但它让药物设计流程中一个长期费时费力的环节,开始变得可计算、可批量、可迭代。
当环肽药物继续向着更小、更复杂、更像药的方向发展时,结构预测模型也必然要从蛋白质世界走向化学世界。AGDIFF 给出的答案很明确:先读懂这张分子图,然后,让三维结构自己慢慢生长出来。
参考文献
Accurate 3D Structure Prediction of Small Cyclic Peptides Containing Non-Canonical Amino Acid Residues Using an All-Atom Diffusion Model with Stereogenic Implementation
Dizhou Wu and Yike Zou
Journal of Chemical Information and Modeling 2026 66 (8), 4398-4408 DOI: https://doi.org/10.1021/acs.jcim.5c03236




